




В табл. 12 представлена генетическая классификация СУИQT: указаны гены, мутации в которых обнаруживают при соответствующих типах заболевания, кодируемые данными генами белки и изменения ионных токов, приводящие к удлинению фаз реполяризации. Следует отметить, что при проведении молекулярно-генетического скрининга больных СУИQT примерно в 25% случаев генетические нарушения не обнаруживают, что позволяет ожидать в дальнейшем выявление новых генетических мутаций, приводящих к возникновению заболевания.
Таблица 12. Молекулярно-генетические типы наследственного синдрома удлинённого интервала QT
| Фенотип | Тип СУИQT | Тип наследования | Мутантный ген | Кодируемый белок | Изменение ионных токов |
| СиндромДжервелла и Ланге–Нильсена | LQT-JLN1 | АР | KCNQ1 | α-субъединица калиевого канала Kv7.1 | Снижение IKs |
| LQT-JLN2 | АР | KCNE1 | MinK— β-субъединица калиевого канала Kv7.1 | Снижение IKs | |
| Синдром Романо–Уорда | LQT1 | АД | KCNQ1 | α-субъединица калиевого канала Kv7.1 | Снижение IKs |
| LQT2 | АД | KCNH2 | α-субъединица калиевого канала Kv11.1 | Снижение IKr | |
| LQT3 | АД | SCN5A | α-субъединица натриевого канала Nav1.5 | Усиление INa | |
| LQT4 | АД | ANK2 | Анкирин B | Снижение INa,K и INcx | |
| LQT5 | АД | KCNE1 | MinK— β-субъединица калиевого канала Kv7.1 | Снижение IKs | |
| LQT6 | АД | KCNE2 | MIRP1 — β-субъединица калиевого канала Kv7.1 | Снижение IKr | |
| Синдром Андерсена–Тавила | LQT7 | АД | KCNJ2 | α-субъединица калиевого канала Kir2.1 | Снижение IK1 |
| Синдром Тимоти | LQT8 | АР | CACNA1c | α-субъединица кальциевого канала L-типа Cav1.2 | Усиление ICa,L |
| Примечание: JLN — синдром Джервелла и Ланге–Нильсена, LQT, СУИQT — синдром удлинённого интервала QT; АД — аутосомно-доминантный; АР — аутосомно-рецессивный |
Описаны следующие фенотипические формы СУИQT: синдром Романо–Уорда (Romano–Ward), синдром Джервелла и Ланге–Нильсена (Jervell and Lange-Nielsen), синдром Андерсена–Тавила (Andresen–Tawil) и синдром Тимоти (Timothy).
Наиболее распространённая форма заболевания с аутосомно-доминантным типом наследования — синдром Романо–Уорда (Romano–Ward), характерными клиническими проявлениями которого являются увеличение продолжительности интервала QT, рецидивирующие синкопальные состояния, чаще всего обусловленные полиморфной желудочковой тахикардией (ЖТ) типа пируэт, и наследственный характер заболевания. Более 90% случаев синдрома Романо–Уорда представлены СУИQT 1-го (СУИQT1), 2-го (СУИQT2) и 3-го (СУИQT3) типов, имеющих особенности клинических и электрокардиографических проявлений (табл. 13, рис. 31).
Таблица 13. Клиническая характеристика основных типов наследственного синдрома удлинённого интервала QT.
| Характеристика | СУИQT1 | СУИQT2 | СУИQT3 |
| Мутантный ген | KCNQ1 | KCNH2 | SCN5A |
| Изменение ионного тока | Снижение IKs | Снижение IKr | Усиление INa |
| Особенности реполяризации на синусовом ритме | Широкая, симметричная волна Т | Низкая амплитуда волны Т, двухфазная волна Т | Удлиненный изоэлектрический сегмент ST |
| Факторы, провоцирующие индукцию TdP | Физическая нагрузка, эмоциональный стресс | Резкий громкий звук, эмоциональный стресс (испуг), резкое начало физической нагрузки | Брадикардия (в покое, во сне) |
| Динамика QTс на нагрузке | Удлинение | Укорочение (нормальная динамика) | Значительное укорочение |
| Эффективность терапии β-адреноблокаторами | Высокая (более 80%) | Умеренная (около 50%) | Низкая (не известна) |
| Динамика QTс при приёме ААП I класса | Нет | Нет | Укорочение |
| Примечание: СУИQT — синдром удлинённого интервала QT; TdP — Torsade de Pointes; ААП —антиаритмический препарат |

Рис. 31. Изменения ЭКГ при различных типах наследственного синдрома удлиненного интервала QT: (А) — широкая гладкая волна T при СУИQT1; (Б) — двухфазная T-волна при СУИQT2; (В) — низкоамплитудная и укороченная T-волна с удлиненным, горизонтальным ST-сегментом при СУИQT3.
СУИQT1 — наиболее распространенный тип синдрома, обусловленный мутацией в гене KCNQ1, кодирующем α-субъединицу калиевого канала, генерирующего ток IKs, который является основным током реполяризации при высокой частоте сердечного ритма. Снижение силы IKs приводит к недосаточному укорочению интервала QT при нарастании частоты сердечных сокращений. По этим причинам больным СУИQT1 характерно возникновение TdP на фоне физической нагрузки (рис. 32) и эмоционального стресса. Особенностью ЭКГ при СУИQT1 является удлиненная и гладкая волна Т (см. рис. 31А).
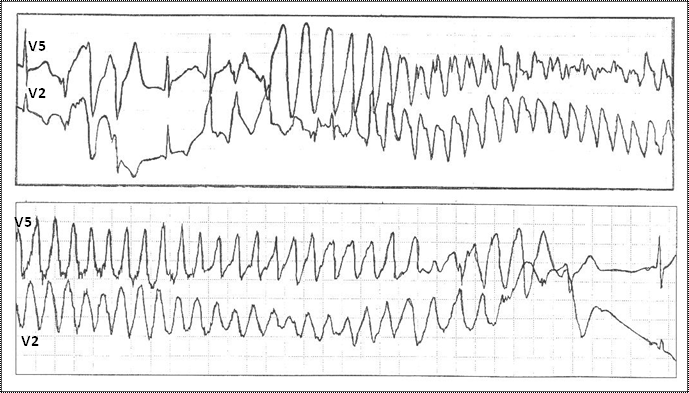
Рис. 32. Развитие пароксизма полиморфной желудочковой тахикардии типа Torsade de Pointes на фоне физической нагрузки у больной синдромом Романо–Уорда (фрагмент непрерывной записи суточного мониторирования ЭКГ по Холтеру).
Причиной СУИQT2 является мутация в гене KCNH2, кодирующем α-субъединицу калиевого канала Kv11.1, генерирующего ток IKr. При СУИQT2 пароксизмы TdP могут возникать как во время нагрузки, так и в покое. Характерным провоцирующим фактором является резкий громкий звук. На ЭКГ больных СУИQT2 регистрируют непротяженную, двухфазную волну Т (см. рис. 31Б).
СУИQT3 является менее распространённой формой заболевания, обусловленной мутацией в гене SCN5A, кодирующем α-субъединицу натриевого канала, что приводит к нарушению инактивации натриевых каналов, продолжающемуся вхождению ионов Na+ в клетку и увеличению продолжительности реполяризации кардиомиоцитов. TdP у больных СУИQT3 возникают на фоне брадикардии, преимущественно во время сна. Физические нагрузки, напротив, переносятся хорошо и сопровождаются укорочением интервала QT. Характерной особенностью ЭКГ у данных больных является удлиненный сегмент ST с отсроченным началом непродолжительной низкоамплитудной волны Т (см. рис. 31В).
Существенно реже встречается аутосомно-рецессивная форма заболевания (синдром Джервелла и Ланге–Нильсена), для которой характерны врождённая нейросенсорная тугоухость, более выраженное увеличение длительности интервала QT и большая частота опасных для жизни желудочковых аритмий. Заболевание обусловлено мутациями в генах KCNQ1 или KCNE2, кодирующих основную и добавочную субъединицы потенциал-зависимых калиевых каналов Kv7.1, приводящими к снижению силы тока IKs.
Синдром Андерсена–Тавила — редкая форма заболевания, при которой удлинение интервала QT сопровождается появлением волны U, пароксизмами как полиморфной желудочковой тахикардии типа TdP, так и двунаправленной желудочковой тахикардии. В 60% случаев заболевание обусловлено мутацией в гене KCNJ2, кодирующем α-субъединицу калиевых каналов аномального входящего выпрямления Kir2.1, генерирующих ток IK1, сила которого снижается. В 40% случаев генетический дефект в настоящее время обнаружить не удаётся. Характерные экстракардиальные проявления заболевания, такие как, аномалии развития костной системы (низкорослость, микрогнатия, большое расстояние между глазницами, низкое расположение ушных раковин, сколиоз, клинодактилиия), гипокалемия и периодический калий-зависимый паралич, присутствуют не у всех больных. Синдром Андерсена–Тавила — заболевание с аутосомно-доминантным типом наследования, однако семейный характер заболевания прослеживается далеко не всегда, в связи с трудностями диагностики, неспецифическими клиническими проявлениями заболевания и неполной пенетрантностью мутантных генов. До 50% случаев заболевания обусловлены мутацией de novo
Синдром Тимоти — крайне редкая форма СУИQT, обусловленная мутацией в гене CACNA1c, кодирующем α-субъединицу кальциевых каналов CaV1.2. При данном синдроме отмечают наиболее выраженное удлинение интервалов QT и QTc (до 700 мс), сопровождающееся крайне высоким риском ВСС (средняя продолжительность жизни составляет 2,5 года). До 60% больных имеет различные врождённые пороки сердца [открытый артериальный проток, тетраду Фалло, открытое овальное окно и дефекты межжелудочковой перегородки] и различные нарушения проводимости (характерны преходящая и постоянная формы АВ блокады II степени с проведением на желудочки 2:1). Среди экстракардиальных проявлений заболевания описаны когнитивные нарушения (задержка психомоторного развития, аутизм), гипогликемия, иммунодефициты, аномалии строения лица (сглаженность носогубной складки, низкое расположение ушных раковин), а также частичное или полное сращение пальцев кистей и стоп (синдактилия). Синдром Тимоти наследуется по аутосомно-доминантному типу, однако подавляющее большинство случаев заболевания обусловлено мутацией de novo.

