




Альдегиды
В ароматических альдегидах альдегидная группа непосредственно связана с бензольным кольцом, в котором могут находиться и другие заместители
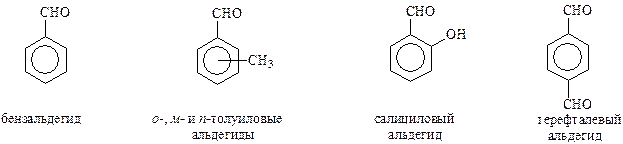
Методы получения
Для получения ароматических альдегидов могут быть использованы все ранее рассмотренные методы: окисление первичных спиртов, восстановление карбоновых кислот, гидролиз гем-дизамещенных галогенпроизводных, пиролиз смешанных солей карбоновых кислот и др. Вместе с тем ароматические альдегиды получают и с использованием специфических методов, которые пока еще не рассматривались. Обращает на себя внимание тот факт, что эти методы базируются на преобразованиях определенных функциональных групп уже находящихся в бензольном кольце.
15.1.1. Превращения арилметанов. В отличие от алифатического ряда, в ароматическом ряду возможна прямая трансформация метильной группы в альдегидную группу в арилметанах. Наиболее просто это достигается парофазным окислением метильной группы в аренах кислородом воздуха в присутствии окиси ванадия или жидкофазным окислением при 40оС двуокисью марганца в 65%-ной серной кислоте

В обоих случаях реакция может идти дальше, поскольку альдегиды легко окисляются до карбоновых кислот. Для предотвращения этого стремятся связать альдегид по мере его образования.
Одним из таких способов является окисление метилпроизводных раствором хромового ангидрида в уксусном ангидриде. В этом случае альдегид по мере образования превращается в гем -диацетат, устойчивый к окислению. Кислотным гидролизом диацетата получают сам альдегид

Другим примером реакции подобного типа является реакция Этара. В этом случае окисление метильной группы осуществляется хлористым хромилом в сероуглероде при 25-45оС. При этом получается молекулярный комплекс, содержащий два эквивалента хлористого хромила. При разложении последнего водой получается альдегид, хромовая кислота и хлористый хром

15.1.1.2. Превращения хлорметильной группы в арилметилгалогенидах. Одними из доступных органических соединений, из которых могут быть получены ароматические альдегиды, являются арилметилхлориды. Превращение хлорметильной группы в альдегидную группу может быть достигнуто несколькими способами. Кроме окислительного гидролиза это реакция Соммле.
В реакции Соммле арилметилгалогенид реагирует с уротропином (гексаметилентетрамином) с образованием четвертичной соли, которая через ряд стадий окисляется в альдегид

В реакцию Соммле не вступают арилметилгалогениды, в молекуле которых хлорметильная группа соседствует с другими заместителями.
15.1.1.3. Восстановление производных карбоновых кислот. Из производных карбоновых кислот для получения альдегидов чаще всего используются хлорангидриды. Восстановление в этом случае можно вести на никелевых или палладиевых катализаторах. Для предупреждения дальнейшего восстановления образовавшегося альдегида в спирт, катализатор несколько «отравляют» обработкой, например, серой в хинолине (метод Розенмунда)
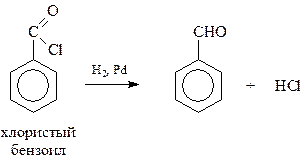
В случае некоторых пространственно затрудненных галогенангидридов необходимость в отравлении катализатора серой отпадает.
Кроме восстановления хлорангидридов кислот по Розенмунду, известны и другие методы восстановления. Считается, что одним из лучших из них является восстановление три- трет -бутоксиалюмогидридом лития при -78оС в растворе диглима

Низкая температура реакции препятствует дальнейшему восстановлению альдегида в спирт.
Следует отметить, что кроме восстановления известны и другие методы превращения галогенацилов в альдегиды (реакции Грундмана, Зона-Мюллера и др.).
Альдегиды могут быть получены и из нитрилов карбоновых кислот. В этом случае нитрил смешивают с безводным хлористым оловом и через смесь пропускают хлористый водород (реакция Стефена). При этом последовательно получается неустойчивый иминохлорид, альдиминовый комплекс и, при гидролизе последнего, альдегид

15.1.1.4. Прямое введение альдегидной группы. Введение альдегидной группы в ароматическое кольцо достигается в условиях реакций Гаттермана-Коха и Реймера-Тиммана. Обе эти реакции относятся к реакциям электрофильного замещения в ароматическом ряду и поэтому они протекают с соблюдением соответствующих закономерностей.
В реакции Гаттермана-Коха на ароматическое соединение (кроме самого бензола и соединений с электроноакцепторными заместителями) действуют смесью хлористого водорода и оксида углерода в присутствии хлористого алюминия и однохлористой меди
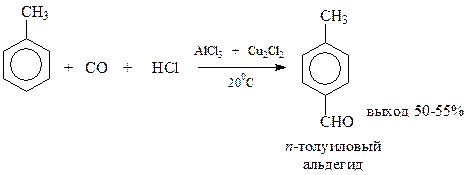
Предполагается, что вначале оксид углерода и хлористоводородная кислота образуют нестойкий хлористый формил, который дальше реагирует по реакции Фриделя-Крафтса

В случае ароматических соединений с электронодонорными заместителями вместо оксида углерода можно использовать синильную кислоту или ее соли (реакция Гаттермана), а в качестве катализатора – хлористый цинк

В реакции Реймера-Тиммана на активное в реакции электрофильного замещения ароматическое соединение действуют хлороформом и щелочью

Имеются основания трактовать реакцию Реймера-Тимана как электрофильное замещение, т.к. при ее осуществлении появляется активный фенолят-ион

А электрофильная частица при этом возникает под действием щелочи из хлороформа

В дихлоркарбене углерод окружен лишь шестью электронами и поэтому он является сильным электрофилом.
В реакции Реймера-Тимана вначале образуется бензальхлорид, который в щелочной среде гидролизуется в альдегид

На участие в рассматриваемой реакции именно фенолят-иона указывает то обстоятельство, что соединения, которые не могут образовать этот ион (например, анизол), не участвуют в реакции Реймера-Тимана.
Химические свойства
Молекулы ароматических альдегидов состоят из бензольного кольца и альдегидной группы. Нетрудно предвидеть, что характерными для подобных соединений будут реакции электрофильного замещения с участием ароматического кольца (альдегидная группа – м -ориентант) и нуклеофильного присоединения по карбонильной группе альдегида. Поскольку указанные реакции рассматривались раньше, ограничимся изучением лишь тех превращений ароматических альдегидов, которые относятся к специфическим, присущим только им.
15.1.2.1. Автоокисление. Как известно, альдегидная группа легко окисляется в карбоксильную группу. В данном случае речь идет о самопроизвольном протекании этого процесса при контакте альдегида с кислородом воздуха, особенно под воздействием света и железа. Налитый тонким слоем бензальдегид при контакте с воздухом уже через несколько часов окисляется в кристаллическую бензойную кислоту.
Установлено, что самоокисление ароматических альдегидов протекает по цепному радикальному механизму. На начальных стадиях процесса – стадии инициирования цепи образуется бензоильный радикал

При развитии цепи образуется перекисный радикал и гидроперекись

Реакция гидроперекиси с альдегидом приводит к кислоте

В пользу приведенного механизма говорит факт присутствия в окисленном бензальдегиде гидроперекиси. Кроме того, известно, что автоокисление предотвращается ингибиторами.
15.1.2.2. Конденсации. При рассмотрении алифатических альдегидов было установлено, что альдегидная группа обуславливает способность соединений этого класса к различным реакциям конденсации: альдольной, кротоновой и др. Оказалось, что отдельные реакции конденсации альдегидов вполне могут быть сведены к реакциям нуклеофильного присоединения по поляризованной карбонильной группе. В реакциях конденсации одна молекула альдегида образует нуклеофильную частицу, а другая молекула альдегида присоединяет этот нуклеофил.
Ароматические альдегиды также вступают в различные реакции конденсации. Своеобразие реакций конденсации с участием ароматических альдегидов заключается в том, что они не могут иметь α -водородных атомов.
Реакция Канницаро. Ароматические альдегиды в присутствии концентрированного водного раствора щелочи вступают в реакцию Канницаро – самоокисления-восстановления с образованием молекулы спирта и карбоновой кислоты
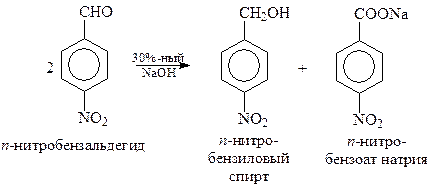
На основе данных кинетических исследований и опытов с мечеными соединениями для реакции Канницаро предложен следующий механизм.
В начальной стадии имеет место нуклеофильное присоединение гидроксила к карбонилу

Образовавшаяся частица поставляет еще один нуклеофил – гидрид-ион – для еще одного нуклеофильного присоединения

При участии в перекрестной реакции Канницаро ароматического альдегида и формальдегида из-за легкой окисляемости алифатического альдегида неизменно получается ароматический спирт и формиат натрия
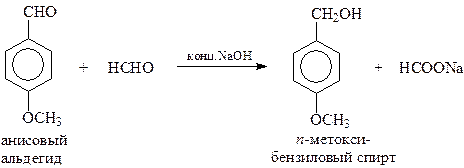
Интересно, что реакция Канницаро может протекать и как внутримолекулярный процесс
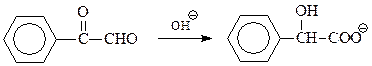
Бензоиновая конденсация. Под действием цианистых калия или натрия ароматические альдегиды вступают в так называемую бензоиновую конденсацию с образованием α -оксикетонов (ацилоинов)

Этот вид конденсации получил свое название по названию продукта конденсации бензальдегида – бензоина.
Предполагается, что бензоиновая конденсация идет по следующей схеме. Первоначально в результате нуклеофильного присоединения цианид-иона к альдегидной группе получается нуклеофильная частица

которая, в свою очередь, вступает в реакцию нуклеофильного присоединения ко второй молекуле альдегида

Продукт присоединения отщепляет цианид-ион и образует бензоин

Уникальность цианид-иона при бензоиновой конденсации вытекает из того, что он обладает достаточной для получения 1 нуклеофильностью, стабилизирует 2 и легко отщепляется от 3 с образованием ацилоина.
Бензоин обладает рядом интересных свойств. Он легко окисляется азотной кислотой в уксуснокислом растворе или сульфатом меди в водном пиридине в дикетон, который называют бензилом

С бензилом связана одна известная перегруппировка. При нагревании с разбавленным раствором едкого натра бензил перегруппировывается в бензиловую кислоту

В ходе бензиловой перегруппировки фенил-анион мигрирует к соседнему углероду. При этом одна карбонильная группа окисляется, а другая – восстанавливается

Бензиловая перегруппировка интересна тем, что в ходе этой реакции 1,2-фенильный сдвиг происходит при щелочном катализе. Между тем, сдвиги такого типа более характерны для реакций, протекающих при кислотном катализе.
Кротоновая конденсация. В результате реакции бензальдегида с ацетоном (недостаток) в среде разбавленной щелочи образуется бензальацетон или дибензальацетон

Хотя кротоновая конденсация с участием ароматических альдегидов и рассматривается обычно как их специфическое свойство, в действительности все происходит в рамках уже известных закономерностей превращений карбонильных соединений.
Как уже отмечалось, ароматические альдегиды лишены α -водородных атомов. Отсюда следует, что они не способны вступать в реакции типа альдольной или кротоновой конденсации. В то же время понятно, что для них вполне возможны перекрестная альдольная или кротоновая конденсации. При этом ароматические альдегиды могут выступать лишь в качестве акцепторов нуклеофилов (карбанионов), образовавшихся из карбонильных соединений с α -водородными атомами: альдегидов, кетонов, сложных эфиров (конденсация по Кляйзену), ангидридов карбоновых кислот (конденсация по Перкину). Образовавшиеся в результате реакции альдоли (β -оксикарбонильные соединения), у которых с β -углеродом связано бензольное кольцо, дегидратируются настолько быстро, что вместо спиртоальдегидов и кетонов получаются ненасыщенные карбонильные соединения.
Рассмотрим в качестве примера механизм конденсации по Кляйзену бензальдегида и ацетона

По аналогичной схеме идет конденсация по Перкину бензальдегида и уксусного ангидрида.
Конденсации с соединениями с подвижными атомами водорода. В реакциях Кляйзена и Перкина ароматические альдегиды конденсируются с соединениями с активными водородами. В этих соединениях водородные атомы становятся активными под влиянием соседней карбонильной группы. Однако водородные атомы могут стать подвижными и по другим причинам. Так, известно, что активными становятся водороды в о- и п -положениях бензольного кольца в аминах и фенолах. Действительно, для ароматических альдегидов известны реакции конденсации с подобными соединениями с образованием соединений трифенилметанового ряда, к которым относятся и многие красители

Кетоны
В ароматических кетонах карбонильная группа связана хотя бы с одним ароматическим радикалом

Способы получения
Ароматические кетоны можно получить на основе тех же реакций, что и алифатические кетоны. К ним относятся реакции окисления (дегидрирования) вторичных спиртов, пиролиз кальциевых или бариевых солей карбоновых кислот, гидролиз гем -дигалогенидов и др. Дальше будут рассмотрены методы, которые не столько оригинальны, сколько часто применяются для получения кетонов определенного строения.
15.2.1.1. Ацилирование. При ацилировании ароматических соединений (реакция Фриделя-Крафтса) получаются кетоны

Эта реакция, как частный случай электрофильного замещения в ароматическом ряду, была рассмотрена раньше (смотри арены). Однако следует обратить внимание на некоторые ее особенности.
Установлено, что ацилироваться способны только ароматические соединения, несущие в кольце электронодонорные заместители. Поскольку сама ацильная группа относится к электроноакцепторным заместителям, то понятно, что две такие группы в бензольное кольцо вводить не удается.
Для ацилирования могут быть использованы карбоновые кислоты, их ангидриды и галогенангидриды. Наибольшей активностью обладают галогенацилы, наименьшей – сами кислоты.
От ацилирующего агента зависит и природа используемого катализатора. Обычно применяется хлорид алюминия, однако в случае ацилирования карбоновыми кислотами приходится использовать более сильные катализаторы – минеральные кислоты. При этом необходимо иметь в виду, что как ацилирующие агенты – ангидриды и галогенангидриды, так и образующиеся кетоны с хлоридом алюминия образуют комплексы. Поэтому катализатор приходится брать в сопоставимых с ацилирующим агентом количествах.
Перегруппировка Фриса. Для получения ароматических кетонов, содержащих в бензольном кольце гидроксильную группу, особенно подходящей является перегруппировка Фриса. Реакция заключается в перегруппировке сложных эфиров фенолов в условиях реакции Фриделя-Крафтса и обычно трактуется как ее внутримолекулярный вариант

На соотношение между образующимися о - и п -изомерами влияет природа растворителя, количество катализатора и температура реакции. Так, м -крезилацетат при 165оС с 95%-ным выходом превращается в 4-метил-3-оксиацетофенон, а при 25оС – в 2-метил-4-оксиацетофенон (выход 85%)

Установлено, что подобное влияние температуры – общая закономерность: при низкой температуре получаются пара -изомеры, а при более высокой температуре – орто -прозводные.
15.2.1.3. Присоединение магнийорганических соединений к нитрилам. Реактивы Гриньяра обладают способностью присоединяться по тройной связи нитрильной группы. При этом первоначально получается соль кетимина, который гидролизуется в кетон
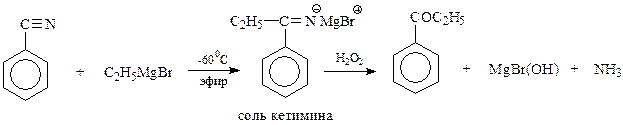
15.2.1.4. Реакция Геша. Активированное бензольное кольцо, содержащее в виде заместителей гидроксильную группу, алкоксигруппы и др., способно конденсироваться (С -алкилирование) с нитрилами в присутствии хлористого водорода и хлористого цинка (Геш, 1915 год). Установлено, что при этом, как и в предыдущем случае, образуется кетимин, гидролизующийся в кетон

Реакцию Геша можно рассматривать и как частный случай реакции Гаттермана – конденсации фенолов и их эфиров с цианистым водородом в присутствии тех же катализаторов.
Химические свойства
Ароматические кетоны, в молекуле которых присутствуют карбонильная группа и бензольное кольцо, вступают в химические реакции, обусловленные указанными функциональными группами. Для карбонильной группы – это нуклеофильное присоединение, замещение карбонильного кислорода и α -водородная активность. Замечено, что ароматические кетоны менее реакционноспособны, чем алифатические кетоны. Так, они не дают производных с бисульфитом натрия. Бензольное кольцо в ароматических кетонах вступает в реакции электрофильного замещения как соединение, несущее электроноакцепторный заместитель.
Рассмотрим некоторые наиболее характерные для ароматических кетонов реакции.
15.2.2.1. Оксимы кетонов, их стереоизомерия и бекмановская перегруппировка. Как известно, карбонильные соединения реагируют с гидроксиламином с образованием оксимов. Эта реакция характерна и для ароматических кетонов

Из строения кетоксима видно, что для некоторых из них возможна геометрическая изомерия. Цис (син)- и транс (анти) – изомеры отличаются по расположению заместителей по двойной связи С=N. Для проявления данного вида геометрической изомерии необходимо участие в реакции с гидроксиламином не симметричных ароматических кетонов

Отметим, что син-изомером принято считать структуру, в которой меньший радикал находится в цис-положении к гидроксильной группе.
Интересно, что син - и анти -кетоксимы способны к взаимным превращениям. Более стабильная анти -форма образуется из син -формы под действием кислот; облучением можно осуществить обратное превращение.
Принадлежность кетоксима к анти - или син -ряду можно установить на основании физических параметров (например, дипольного момента) или химическим путем. Покажем это на примере упомянутых выше фенил- п - метоксифенилкетоксимов.
Известно (Бекман, 1886 год), что оксимы под действием кислот, кислот Льюиса или PCl5 превращаются в замещенные амиды кислот

При бекмановской перегруппировке происходит обмен местами гидроксильной и фенильной групп и кетонизация промежуточно образующегося енольного производного. Предполагается, что бекмановская перегруппировка оксимов протекает по схеме, предусматривающей 1,2-фенильный сдвиг к электронодефицитному атому азота.
При гидролизе амиды кислот дают соответствующие карбоновые кислоты и амины
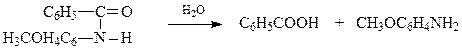
Оказалось, что стереоизомерные оксимы в результате бекмановской перегруппировки с последующим гидролизом образовавшихся амидов дают различные карбоновые кислоты и амины
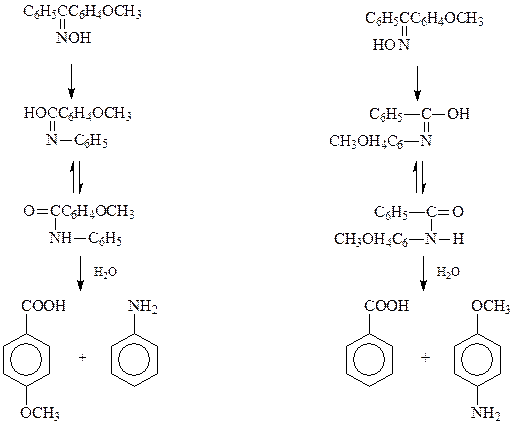
По составу продуктов указанных превращений возможно однозначно отнести соединение к анти - или син -кетоксиму.
15.2.2.2. Реакции α -водородных атомов. Ароматические кетоны способны к многочисленным превращениям с участием α -водородных атомов. Это знакомые уже по алифатическому ряду реакции галогенирования, кротоновой конденсации, сложноэфирной конденсации, конденсации с ангидридами кислот по Меервейну и др.
В качестве примера рассмотрим реакцию Манниха в приложении к ароматическим кетонам. Как известно, эта реакция заключается во взаимодействии кетона с формальдегидом и гидрохлоридом вторичного амина с образованием, так называемых оснований Манниха

Соль основания Манниха неустойчива и при нагревании легко отщепляет гидрохлорид диметиламина

Карбоновые кислоты
Молекула ароматических карбоновых кислот включает такие обязательные структурные элементы как бензольное кольцо, в том числе замещенное, и карбоксильную (карбоксильные) группу. Количество карбоксильных групп определяет основность кислот.
Ароматические карбоновые кислоты обычно имеют свои тривиальные названия, указывающие на их природный источник или характерное свойство. При необходимости их рассматривают как производные более простых кислот. Ниже представлены наиболее часто встречающиеся представители ароматических карбоновых кислот

Методы получения
Карбоновые кислоты рассматриваемого ряда могут быть получены всеми общими способами, которые используются при синтезе алифатических кислот: окисление первичных спиртов и альдегидов, гидролиз тригалогенпроизводных, карбоксилирование натрий - и литийарилов, через магнийорганический синтез, с использованием галоформной реакции и др. Однако разработаны и некоторые специфические методы. При получении ароматических карбоновых кислот чаще всего используются следующие методы.
15.3.1.1. Окисление ароматических соединений. При крупномасштабном получении карбоновых кислот различные варианты окисления используются гораздо чаще, чем в алифатическом ряду. При этом окислению подвергаются не только альдегиды и первичные спирты, но и многие другие классы органических соединений. Сказанное может быть подтверждено следующими примерами.
При окислении арены теряют свой алкильный заместитель и, независимо от его величины и строения, взамен получают карбоксил

Для окисления используют щелочной раствор перманганата калия, хромовую кислоту, азотную кислоту, кислород воздуха в присутствии катализаторов (например, солей марганца).
Кроме бензойной кислоты в промышленности окислением получают и двухосновную кислоту – терефталевую, используемую при синтезе полиэфира в реакции с этиленгликолем (лавсан, терилен)

Несмотря на кажущуюся простоту, указанная реакция оказалась трудной в осуществлении. После окисления одной метильной группы в п -ксилоле, возникшая карбоксильная группа экранирует второй метил и затрудняет его окисление. Однако были найдены условия окисления, позволившие преодолеть указанные затруднения: окислитель – кислород воздуха, растворитель – уксусная кислота, катализатор – соли марганца и кобальта, сокатализатор соединения брома. Окисление проводится при 190оС и давлении 10 атм.
Окислением соответствующих углеводородов кроме терефталевой кислоты получают и не менее важные в производстве полимеров о -фталевый ангидрид (глифталевые смолы) из нафталина и пиромеллитовый диангидрид (полиэфирные смолы) из дурола.
Окисление нафталина (или о -ксилола) проводится кислородом воздуха при высокой температуре (500оС) в присутствии пятиокиси ванадия. В близких условиях окисляют и дурол

Наконец, отметим, что окислению могут быть подвергнуты и ароматические кетоны, которые окисляются даже легче, чем углеводороды. Окисление обычно проводят с помощью гипохлоритов, хотя могут быть использованы и другие окислители

15.3.1.2. Гидролиз нитрилов. Эта реакция, используемая и в алифатическом ряду, находит применение и при синтезе ароматических карбоновых кислот

В отличие от алифатического ряда, в ароматическом ряду гораздо шире возможности получения самих нитрилов кислот. Так бензонитрил может быть получен не только нуклеофильным замещением ароматически связанного галогена, но и сплавлением сульфонатов с цианистыми солями

Для этой цели могут быть использованы и диазосоединения

15.3.1.3. Ацилирование по Фриделю-Крафтсу. Реакция электрофильного замещения в ароматическом ряду позволяет вводить в бензольное кольцо карбоксильную группу непосредственно. Для этого в качестве ацилирующего агента используется фосген или хлоругольный эфир

15.3.1.4. Функционализация ароматических соединений. Среди ароматических карбоновых кислот, встречающихся в природе или синтезируемых и используемых при получении красителей, лекарственных препаратов и других областях применения, встречаются их замещенные представители. Общий подход к синтезу таких кислот предусматривает либо введение функциональной группы в молекулу ароматической карбоновой кислоты, либо создание карбоксильной функции в бензольном кольце, уже несущем определенную функциональную группу.
Галогензамещенные бензойные кислоты получают, используя оба подхода. Галоген вводится в бензольное кольцо прямым галогенированием (хлорирование бензойной кислоты в присутствии хлорида железа), либо с использованием реакции Зандмейера исходя из доступных аминокислот через соли диазония

Нитробензойные кислоты с м -положением нитрогруппы по отношению к карбоксилу, являются продуктами нитрования ароматических карбоновых кислот

Введение карбоксила в молекулу ароматических нитросоединений каким-нибудь из известных методов – путь получения о - и п -нитрокарбоновых кислот

Сульфобензойные кислоты. Среди кислот этого ряда применение в пищевой промышленности находит натриевая соль о -сульфобензойной кислоты – сахарин. Это соединение в 500 раз слаще сахара. Схема получения сахарина иллюстрирует подходы, используемые при синтезе сульфобензойных кислот
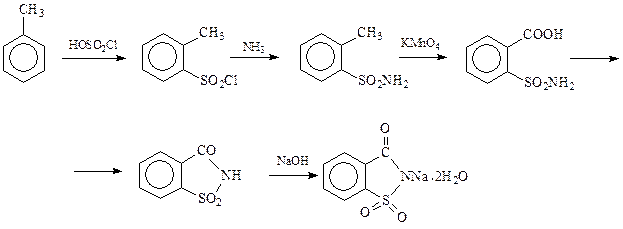
Оксикарбоновые кислоты. Рассмотрим получение ароматических оксикислот на примере некоторых известных их представителей.
В медицине находят применение салициловокислый натрий (лечение суставного ревматизма), ацетат салициловой кислоты – аспирин (жаропонижающее и аналгетическое средство), фениловый эфир салициловой кислоты – салол (средство для дезинфекции кишечника). Салициловую кислоту обычно синтезируют по реакции Кольбе-Шмидта исходя из фенолята натрия

В природе широко распространена триоксибензойная кислота – галловая кислота, в свободном виде или в виде некоторых своих производных. В лаборатории ее можно синтезировать по следующей схеме
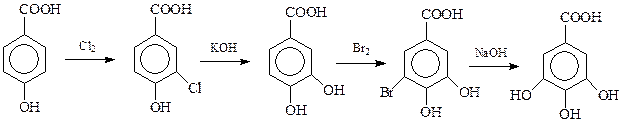
Аминокислоты с различным расположением аминогруппы и карбоксила могут быть получены восстановлением соответствующих нитробензойных кислот. Кроме того, наиболее известная из них – антраниловая кислота синтезируется исходя из фталимида расщеплением по Гофману

Некоторые производные п -аминобензойной кислоты обладают анестизирующим действием и широко применяются в медицине. К ним относятся этиловый эфир упомянутой кислоты – анестезин и хлористоводородная соль диэтиламиноэтилового эфира – новокаин

Дикарбоновые кислоты, особенно о -фталевая и терефталевая, широко применяются при получении полимеров полиэфирного типа. Как уже упоминалось, о -фталевая кислота получается окислением о -ксилола или нафталина.
Терефталевая кислота также может быть получена окислением п -ксилола. Вместе с тем разработаны и другие методы ее получения. К терефталевой кислоте ведет диспропорционирование калиевой соли бензойной кислоты при 340оС и давлении 300 атм. в присутствии бикарбоната калия

Терефталевая кислота является также продуктом изомеризации калиевой соли фталевой кислоты при 400оС в атмосфере СО2 в присутствии фталатов цинка и кадмия

Химические свойства
В соответствии со своим строением ароматические карбоновые кислоты обнаруживают ожидаемые химические свойства. За счет бензольного кольца они вступают в реакции электрофильного замещения. При этом под влиянием карбоксила они реагируют труднее, чем не замещенное соединение, а новый заместитель в результате реакции оказывается в м -положении к карбоксилу.
За счет карбоксильной группы ароматические карбоновые кислоты образуют соли, сложные эфиры, ангидриды и галогенангидриды, амиды и нитрилы.
15.3.2.1. Кислотность. Бензольное кольцо лишь незначительно влияет на кислотность ароматических карбоновых кислот. Для сравнения приведем показатели кислотности рКа (напоминаем, что это отрицательный логарифм константы кислотности Ка; чем меньше значение рКа, тем выше кислотность) ряда карбоновых кислот

Из этих данных видно, что бензольное кольцо действительно слабо влияет на кислотность. Обусловлено это тем, что из двух механизмов передачи электронных эффектов – индуктивного и резонансного, в случае ароматических карбоновых кислот мезомерный эффект не может проявить себя. По валентным соображениям в случае ароматических карбоновых кислот нельзя представить себе резонаносные структуры, в которых заряд карбоксилатной группы переходил бы на ароматическое кольцо.
В тоже время заместители в бензольном кольце сообщают ему положительный или отрицательный индукционный эффект. Этим объясняется наблюдаемая кислотность п -метокси- и п -нитробензойных кислот.
На кислотность ароматических карбоновых кислот могут повлиять и другие факторы, например, водородные связи. Из-за стабилизации салицилат-иона внутримолекулярными водородными связями салициловая кислота проявляет большую кислотность, по сравнению с о -метоксибензойной кислотой, в которой анион не может быть стабилизирован внутримолекулярной водородной связью


По вполне понятной причине двухосновные кислоты имеют две константы кислотности
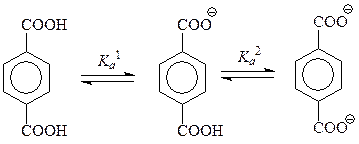
При этом Ка 1› Ка 2, т.к. ионизация второй карбоксильной группы протекает труднее, чем первой – приходится удалять протон не от нейтральной частицы, а от аниона.
15.3.2.2. Действие нагревания. Действие нагревания на ароматические карбоновые кислоты бывает направлено на карбоксильную группу. При термолизе кислот происходит декарбоксилирование или дегидратация с участием двух карбоксилов.
В случае двухосновных ароматических карбоновых кислот дегидратация особенно характерна для тех их представителей, в молекуле которых карбоксилы расположены по соседству (о- изомеры). Так, при нагревании до относительно невысокой температуры 200оС о -фталевая кислота теряет молекулу воды и превращается в свой ангидрид
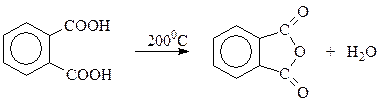
Ангидриды ароматических карбоновых кислот вступают во все реакции, характерные для подобных соединений, и в общем случае образуют другие производные кислот: сложные эфиры, амиды и др. Своеобразие рассматриваемых ангидридов заключается в том, что их превращения приводят к кислым (неполным) производным ароматических карбоновых кислот, которые трудно получить другими методами
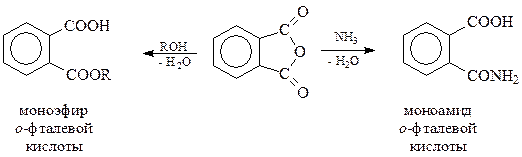
Интересно, что при нагревании моноамидов имеет место их дегидратация с образованием соединений (имидов), в молекуле которых два карбоксила связаны с остатком аммиака

Имиды ароматических карбоновых кислот под влиянием двух ацильных остатков становятся настолько кислыми (Ка =5.10-9 для фталимида), что приобретают способность растворятся в холодном спиртовом растворе КОН
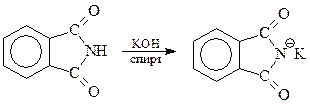
Подобные соли можно использовать как источник нуклеофильной частицы в реакциях нуклеофильного замещения с участием галогеналкилов. Продукт замещения – алкилированный при азоте имид – после гидролиза дает чистый первичный амин

Такой путь синтеза первичных аминов, не загрязненных вторичными и третичными аминами, носит название синтеза чистых первичных аминов по Габриэлю.
15.3.2.3. Участие в реакциях поликонденсации. Как и вообще карбоновые кислоты, ароматические кислоты при взаимодействии со спиртами образуют сложные эфиры. Особый интерес представляют случаи участия в этой реакции двухосновных кислот и двух- и более атомных спиртов

Образовавшийся моноэфир, благодаря наличию свободных карбоксильной и гидроксильной групп, сохраняет способность к дальнейшей этерификации. По этой причине обычная реакция этерификации в случае участия в реакции двухосновных кислот и двухатомных спиртов приводит к полиэфирам

Лавсан используется для изготовления пленок и нитей и тканей из них, обладающих прекрасными потребительскими свойствами.
Не меньший интерес представляют полиэфиры, полученные с участием в поликонденсации о -фталевой кислоты (ангидрида) и глицерина
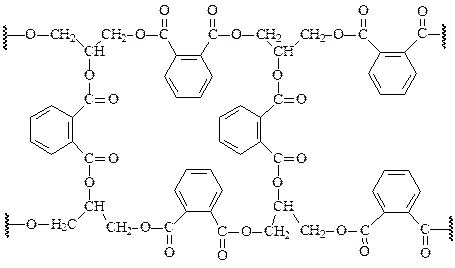
Полиэфиры этого типа – глифталевые смолы – широко используются при получении высококачественных лаков (глифталевые лаки).
Известно, что при поликонденсации используется и пиромеллитовая кислота или ее ангидрид. В этом случае получаются термостойкие полиэфиры.

