




Гидроксильная группа относится к числу наиболее сильных заместителей первого рода (+ М -эффект). Поэтому фенолы легко вступают в реакции электрофильного замещения. При этом новые заместители направляются в о - и п -положения. Активирующее влияние гидроксила в фенолах выражено настолько сильно, что реакцию трудно бывает остановить на стадии монозамещения. Для этого приходится осуществлять тщательный подбор условий проведения реакции. Иногда удается ослабить активирующее влияние гидроксила его трансформацией во что-нибудь менее активное, например, в простую эфирную группу. Защита функциональной группы в этом случае может быть снята реакцией с иодистым водородом.
14.1.2.5. Галогенирование. При взаимодействии фенолов с галогенами реакция идет даже без катализатора с замещением всех о - и п -водородных атомов бензольного кольца. Например, фенол быстро реагирует с бромом в водной среде с образованием 2,4,6-трибромфенола

При избытке брома реакция идет даже дальше. При этом получается пентабромдиенон (Бенедикт, 1879 г.)
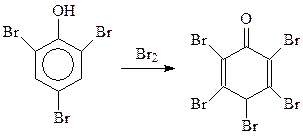
Тщательным подбором условий бромирования в бензольное кольцо можно вводить один или два атома галогена

14.1.2.6. Нитрование. Фенол нитруется даже без применения нитрующей смеси. При этом с помощью концентрированной азотной кислоты в ароматическое ядро фенола можно ввести сразу три нитрогруппы с получением 2,4,6-тринитрофенола (пикриновой кислоты)

Разбавленная азотная кислота позволяет получить продукты мононитрования фенола

Прямое нитрование фенола в мононитрофенолы – едва ли удобный метод их получения из-за того, что выход соединений оставляет желать лучшего. Правда, смесь о - и п -нитрофенолов достаточно легко разделяется перегонкой с водяным паром. о -Нитрофенол образует внутримолекулярную водородную связь и летуч с паром, а п -изомер ассоциируется с молекулами воды также через водородную связь и менее летуч
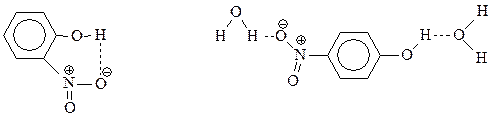
14.1.2.7. Сульфирование. Под действием концентрированной серной кислоты фенол сульфируется даже при комнатной температуре с образованием примерно равных количеств о - и п -оксибензолсульфокислот
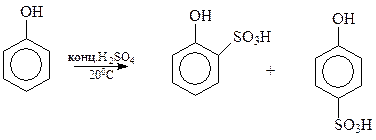
Оказалось, что результат сульфирования сильно зависит от температуры процесса. При 1000С состав продуктов сульфирования определяется накоплением более термодинамически устойчивого n -изомера (продукт термодинамического контроля), при 200С происходит накопление продуктов в соответствии с скоростями их образования (кинетический контроль)
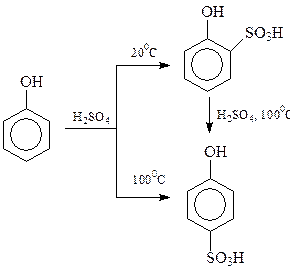
14.1.2.8. Алкилирование. Фенолы легко вступают в реакции алкилирования и давно уже соответствующие реакции используются для получения алкилфенолов в промышленности. Это приходится подчеркивать, потому что в научной литературе почему-то сложилось мнение, что «выходы в этой реакции часто бывают небольшими».
В качестве примера приведем реакцию алкилирования фенола изобутиленом в присутствии серной кислоты (толуолсульфокислоты, катионообменных смол)

Во многих реакциях алкилирования алкильная группа направляется в наименее пространственно экранированное п -положение бензольного кольца.
Однако когда оно занято, даже такие объемистые группы как трет -бутильный радикал, занимают свободные о -положения. Как уже упоминалось, эта реакция используется при синтезе ионола

Попутно отметим, что в ионоле две трет -бутильные группы настолько экранируют гидроксильную группу, что соединение теряет фенольные свойства и не реагирует с щелочами, щелочными металлами, уксусным ангидридом. В то же время экранированные фенолы приобретают другие ценные свойства, которые будут обсуждены позднее.
При алкилировании фенолов в качестве алкилирующих агентов могут быть использованы алкены, спирты, галогенпроизводные. Реакцию катализируют минеральные кислоты и кислоты Льюиса.
Алкилирование фенолов является частным случаем реакции электрофильного замещения в ароматическом ряду. Электрофильной частицей в данном случае выступают карбокатионы. Поэтому, как и в других реакциях с участием карбокатионов, при алкилировании фенолов имеет место их перегруппировка. В результате фенол получает не тот алкил, который был в алкилирующем агенте

14.1.2.9. Ацилирование. Ацилированию фенолов с использованием в качестве катализаторов кислот Льюиса мешает реакция фенолов с катализатором
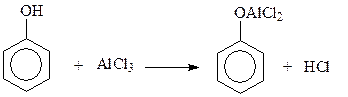
Поэтому прямое ацилировние фенолов практически не используется в синтезах. В случае необходимости введения кислотного остатка в бензольное ядро фенолов приходится защищать гидроксильную группу

Следует отметить, что ацилфенолы лучше всего получать, по-видимому, кляйзенской перегруппировкой сложных эфиров фенолов.
Один из наиболее широко используемых кислотно-основных индикаторов – фенолфталеин – получается «ацилированием» фенола фталевым ангидридом в присутствии серной кислоты

Из строения фенолфталеина видно, что эта реакция идет своеобразно и может быть отнесена к реакциям ацилирования лишь по формальным признакам.
Бесцветный фенолфталеин при рН>9 (в щелочных растворах) теряет оба протона с образованием окрашенного в интенсивный красный цвет дианиона

При рН ниже 8.5 идет обратная реакция превращения дианиона в бесцветную форму.
14.1.2.10. Нитрозирование. Фенолы достаточно активны, чтобы прореагировать с нитрозоний-катионом NO+, являющимся слабым электрофилом

Нитрозогруппа в фенолах может быть доокислена до нитрогруппы при помощи азотной кислоты.
14.1.2.11. Азосочетание. Значительной активностью фенолов в реакциях электрофильного замещения объясняется и их реакция с солями диазония. Известно, что диазоний-катион относится к слабым электрофилам и способен реагировать лишь с очень активными ароматическими соединениями, содержащими в ядре сильные электронодонорные группы, в том числе – гидроксил

Эта реакция азосочетания осуществляется обычно в слабощелочной среде, поскольку в этом случае в реакционной среде возрастает концентрация фенолят-аниона. Наличие отрицательного заряда на атоме кислорода делает его более сильным донором, а фенолят-анион – более реакционноспособным в реакциях электрофильного замещения

14.1.2.12. Реакция Кольбе-Шмидта. При взаимодействии фенолята натрия с диоксидом углерода при 1250С и 4-7 атм. (Кольбе, 1859 г., Шмидт, 1885 г.) происходит карбоксилирование бензольного кольца преимущественно в о -положение

Диоксид углерода относится к числу слабых электрофилов

Чтобы этот слабый электрофил мог участвовать в реакции электрофильного замещения, требуется очень активное бензольное кольцо, такое как в фенолят-анионе.
Реакцию Кольбе-Шмидта вполне можно трактовать как реакцию электрофильного замещения с участием слабого электрофила. Однако при этом нелегко объяснить склонность этой реакции к предпочтительному образованию именно о -оксибензойной кислоты. Этим вызваны, по-видимому, попытки рассматривать реакцию Кольбе-Шмидта как реакцию присоединения карбаниона к диоксиду углерода

В таком виде реакция напоминает карбонизацию реактива Гриньяра, в результате которой получаются карбоновые кислоты.
Для объяснения преимущественного о -замещения в реакции Кольбе-Шмидта предлагался также механизм, аналогичный механизму О -алкилирования фенолов
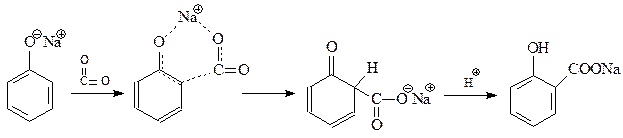
Благодаря реакции Кольбе-Шмидта стала доступной салициловая кислота, на основе которой получают такие широко известные лекарственные препараты как метилсалицилат, аспирин, салол

14.1.2.13. Реакция Реймера-Тимана. Реакция Реймера-Тимана заключается во взаимодействии фенола хлороформом в водной щелочной среде. В результате этой реакции в бензольное ядро вводится альдегидная группа в о -положение к гидроксилу

Предполагается, что реакция Реймера-Тимана – еще один случай электрофильного замещения с участием фенола и необычного электрофила, в качестве которого выступает дихлоркарбен. Он образуется в результате взаимодействия хлороформа с щелочью

В дихлоркарбене углерод окружен лишь секстетом электронов и поэтому является сильным электрофилом. Реакцию дихлоркарбена с фенолят-анионом можно представить следующей схемой
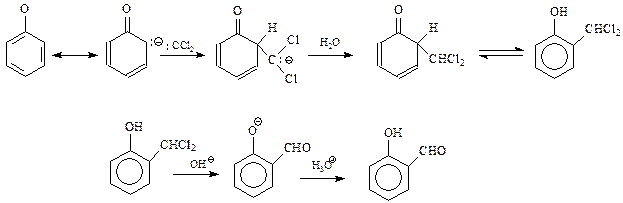
14.1.2.14. Фенол-формальдегидные смолы. Реакцию фенола с формальдегидом с образованием ценных полимеров установил Лео Бакеланд, поэтомуфенол-формальдегидные смолы часто называют бакелитами.
Образование полимера из фенола и формальдегида складывается из двух стадий. На первой стадии в результате взаимодействия фенола с формальдегидом образуется смесь фенолов, содержащих в о - и п -положениях оксиметильные группы. Интересно, что рассматриваемая стадия конденсации катализируется и кислотами и щелочами
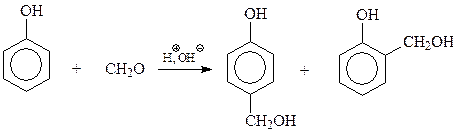
Кислотный катализатор ускоряет конденсацию за счет создания более активного электрофила

При щелочном катализе активируется фенольная компонента в результате образования более активного, чем фенол, фенокси-аниона

Сама первая стадия конденсации фенола с формальдегидом допускает ее трактовку как реакции электрофильного ароматического замещения, и как нуклеофильного присоединения фенокси-аниона к карбонильной группе формальдегида с образованием фенолоспиртов


В дальнейшем происходит присоединение фенокси-аниона к продукту дегидратации фенолоспиртов под названием хинонметид


В результате этой реакции происходит «сшивание» двух молекул фенолоспиртов через метиленовый мостик. Полученный при этом продукт – еще не полимер. Бакелит получается при протекании вышеприведенной реакции с участием фенолоспиртов, замещенных во всех о - и п -положениях на группу СН2ОН

Хотя фенолформальдегидные смолы относятся к числу старейших синтетических полимеров, они и в наши дни производятся и используются в огромных количествах. Примерно половина производимого в промышленности фенола используется при получении фенолформальдегидных смол.
14.1.2.15. Гидрирование. Бензольное кольцо в фенолах может быть прогидрировано с получением циклогексанолов. Обычно образуется смесь циклогексанолов, циклогексанонов и циклогексанов. В качестве катализаторов гидрирования фенолов чаще всего используются платина и никель.
14.1.2.16. Окисление. Появление гидроксильной группы в бензольном ядре делает его более уязвимым к действию окислителей. Даже при хранении фенол может окислиться кислородом воздуха с образованием окрашенных веществ. Попутно отметим, что введение в бензольное кольцо фенолов электроноакцепторных заместителей повышает его устойчивость к действию окислителей.
Рассмотрим несколько примеров окисления фенола разными окислителями.
При окислении фенола перекисью водорода в присутствии железного катализатора получается пирокатехин с небольшим выходом. Хромовая смесь направляет окисление фенола в сторону п -бензохинона

Наибольший интерес представляет рассмотрение окисления пространственно затрудненных (экранированных) фенолов с объемистыми алкильными заместителями в о - и п -положениях к гидроксилу.
При окислении таких фенолов кислородом воздуха, окисью серебра, феррицианидом калия и др. образуются ароксилы, представляющие собой свободные радикалы
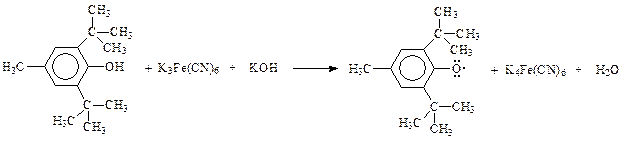
Такого типа арокси (фенокси) радикалы настолько стабильны, что в отсутствии воздуха или других активных реагентов могут существовать в неизменном виде неопределенно долго.
Стабилизация радикала, образовавшегося при окислении экранированного фенола, обусловлена рассредоточением неспаренного электрона по всей ароматической системе. Это хорошо видно, если радикал представить как резонансный гибрид возможных канонических структур

Роль алкильных заместителей в о - и п -положениях бензольного кольца радикала заключается в стерическом препятствовании их рекомбинации – слиянию двух радикалов между собой как через кислород, так и с участием кольца.
Благодаря своим уникальным свойствам, экранированные фенолы проявили себя хорошими ингибиторами, в частности, антиоксидантами. Они «подставляют» себя под удар радикала – участника цепной реакции. Образующийся при этом стабильный и малоактивный радикал не способен продолжать цепь
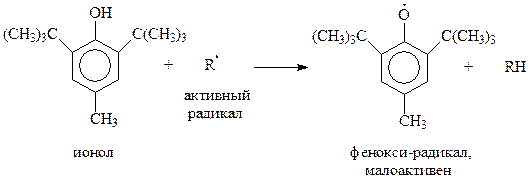
Многоатомные фенолы
Известны многочисленные фенолы, содержащие в бензольном кольце несколько, вплоть до шести, гидроксильных групп. Они как таковые или в виде производных широко распространены в природе, особенно в растительном мире.
Для получения ди- и полифенолов применяются общие методы синтеза фенолов. Однако нередко прибегают и к специфическим методам.
14.2.1. Методы получения. Рассмотрим методы получения важнейших представителей многоатомных фенолов.
Пирокатехин может быть получен гидролизом о -дихлорбензола (водный раствор NaOH, 2000С, катализатор – соли меди)
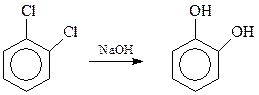
или с использованием реакции щелочного плава из динатриевой соли о -бензолдисульфокислоты

Интересно, что при температуре щелочного плава – свыше 3000С в результате реакции может получиться вместо ожидаемого пирокатехина изомерный ему резорцин. Предполагается, что изомеризация происходит через промежуточное образование дегидробензола
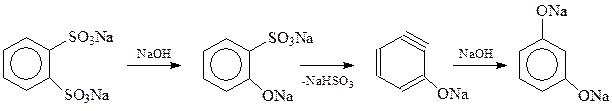
Аналогично изомеризуется и п -бензолдисульфокислота.
Движущей силой указанных превращений является, по-видимому, накопление при высоких температурах наиболее устойчивого из двухатомных фенолов – резорцина.
Источником пирокатехина могут послужить и вещества природного происхождения. В некоторых растениях содержится протокатехиновая кислота, декарбоксилирование которой приводит к пирокатехину

Известен метод получения пирокатехина из салицилового альдегида. При окислении альдегида перекисью водорода в щелочном растворе происходит замещение альдегидной группы на гидроксил

Резорцин получается при щелочном плаве м -бензолдисульфокислоты

Гидрохинон может быть получен, как и другие двухатомные фенолы, из соответствующих дигалоген- и дисульфопроизводных бензола. В то же время для этого фенола известен и свой специфический метод, заключающийся в окислении анилина хромовой кислотой до п -хинона и восстановлении последнего в гидрохинон. Предполагается, что реакция идет по следующей схеме
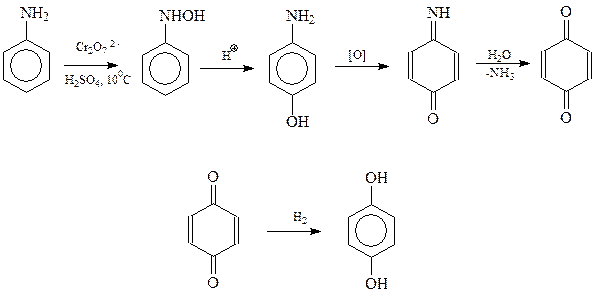
Пирогаллол. Доступным исходным соединением для получения пирогаллола является природная галловая кислота, выделяемая из продуктов гидролиза дубильных веществ. Галловая кислота при нагревании легко декарбоксилируется

Флюроглюцин в виде своих производных широко распространен в растительном мире. Из нескольких синтетических методов его получения наиболее экстравагантным выглядит метод, основанный на превращениях триаминобензойной кислоты

Гексаоксибензол. Этот фенол, содержащий максимально возможное число гидроксилов в бензольном кольце и обладающий симметричной и совершенной структурой, не мог не привлечь к себе внимания химиков. Для него предложены несколько неочевидных методов получения.
Установлено, что простейший диальдегид – глиоксаль в водном растворе сульфита и бикарбоната натрия при пропускании через раствор воздуха превращается в гексаоксибензол. Этот фенол получают также при подкислении продукта взаимодействия металлического калия и оксида углерода
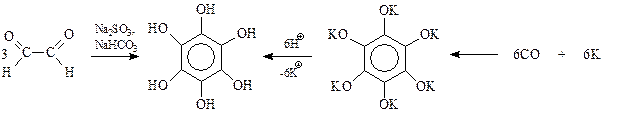
Химические свойства
Многоатомным фенолам присущи свойства, характерные и для одноатомных фенолов. В то же время почти у каждого из них встречаются свои особенности.
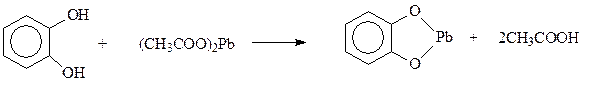
Кислотность. У двухатомных фенолов кислотные свойства выражены сильнее, поэтому они способны образовать соли не только с щелочными металлами
Реакции по бензольному кольцу. В реакции электрофильного замещения двухатомные фенолы вступают много легче, чем фенол. В кольцо двухатомных фенолов удается ввести два и даже три новых заместителя даже в мягких условиях. При электрофильном замещении с участием резорцина гидроксилы действуют согласованно


Окисление. Двухатомные фенолы с гидроксилами в о - и п -положениях легко окисляются и поэтому являются сильными восстановителями. Продуктами окисления пирокатехина и гидрохинона являются соответствующие хиноны

Резорцин также способен окисляться, однако по структурным соображениям не может образовать хинон.
Особенности многоатомных фенолов. Как уже отмечалось, почти каждый из многоатомных фенолов, участвуя в химических реакциях, проявляет особенности. Так, резорцин – единственный из фенолов, способный восстанавливаться водородом в момент выделения. Установлено, что восстанавливается при этом енольная форма резорцина

Точно также гидрохинон – единственный из фенолов, участвующий в реакции Дильса-Альдера как диен (Куксон, 1955 г.)

Не менее своеобразно окисляется шестиатомный фенол. При окислении кислородом воздуха он вначале превращается в тетраоксихинон. В дальнейшем хинон образует диоксихинон - родизоновую кислоту. Продуктом исчерпывающего окисления последней азотной кислотой является трихиноил (гексакетоциклогексан)

Хиноны
Хиноны относятся к сопряженным циклическим дикетонам. У них с фенолами тесные взаимоотношения: фенолы и хиноны легко превращаются друг в друга.
Простейший и наиболее распространенный хинон был получен Воскресенским (1838 г.) окислением хинной кислоты (1,3,4,5-тетраоксигексагидробензойная кислота) двуокисью марганца в серной кислоте
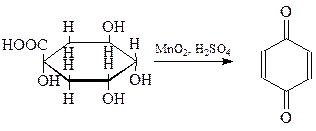
Свое название хинон получил от названия хинной кислоты.
Хиноны подразделяются на 1,2- и 1,4-хиноны или о - и п -хиноны. Как уже отмечалось, 1,3-хиноны не могут существовать по структурным соображениям. Интересно, хиноны различимы по цвету кристаллов: п -хиноны в большинстве случаев бывают желтыми, а о -хиноны – оранжевыми или красными.
Основные методы получения хинонов уже рассматривались – это окисление двухатомных фенолов и окисление анилина хромовой кислотой.
Хиноны обладают высокой реакционной способностью. Направления их превращений предсказуемы: они реагируют как кетоны и одновременно по карбонильной группе и двойным связям. Для них характерны реакции 1,4- и 1,6-присоединения

Хинон как кетон вступает в реакцию с гидроксиламином с образованием моно- и диоксима
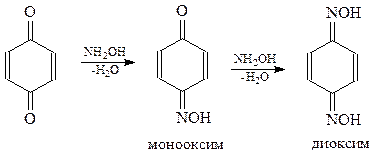
Хиноны вступают в многочисленные реакции присоединения.
Присоединение брома происходит по 3,4-положению п -хинона. Образующийся при этом дибромид легко отщепляет молекулу бромистого водорода и дает замещенный хинон

Аналогично протекает присоединение диенов. В реакции Дильса-Альдера хиноны выступают как диенофилы

Однако хиноны и сами могут реагировать по 1,3-положению подобно диенам с сопряженными двойными связями. Примером такой реакции является присоединение к хинонам галогенводородных кислот

Аналогично присоединяется к хинону и уксусный ангидрид.
Примером реакции 1,6-присоединения является присоединение водорода с образованием двухатомных фенолов

Ароматические спирты
Ароматические спирты классифицируются в зависимости от взаимного расположения гидроксила и бензольного кольца.
Спирты с α -положением гидроксила содержат гидроксил у углерода боковой цепи, связанного с бензольным кольцом. Их получают с использованием уже известных методов синтеза спиртов.
Галогенпроизводные с α -положением атома галогена легко могут быть получены галогенированием алкилбензолов при освещении ультрафиолетом. Такие галогенпроизводные легко гидролизуются даже при кратковременном нагревании с раствором соды

Другой часто используемый метод – восстановление ароматических альдегидов
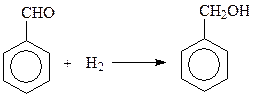
Восстановление альдегидов можно вести и путем вовлечения их в перекрестную реакцию Канницаро когда одним из альдегидов выступает формальдегид. При этом из двух альдегидов формальдегид неизменно окисляется, а ароматический альдегид – восстанавливается

Нередко для получения ароматических спиртов с α -расположением гидроксила исходят из соответствующих сложных эфиров, легко восстанавливающихся атомарным водородом по Буво и Блану (металлический натрий и спирт)

Спирты с β -расположением гидроксила не могут быть получены из соответствующих галогенпроизводных, которые легче подвергаются дегидрогалогенированию, чем гидролизу. Между тем, β -фенилэтиловый спирт широко применяется в парфюмерии («розовое масло»). Поэтому были разработаны эффективные методы его синтеза реакцией фенилмагнийбромида с окисью этилена

В последующем оказалось, что реакцию можно существенно упростить, сведя ее к реакции Фриделя-Крафтса

Представитель ароматических спиртов с γ -положением гидроксила - γ -фенилпропиловый спирт – легче всего синтезировать восстановлением коричной кислоты

Сама кислота получается из бензальдегида и уксусного ангидрида при их конденсации по Перкину.
γ -Фенилпропиловый спирт обладает запахом гиацинтов и применяется в парфюмерии.
Ароматические спирты по своим свойствам повторяют свойства спиртов алифатического ряда.
К особенностям ароматических спиртов можно отнести ту легкость, с которой происходит замещение гидроксила на галоген у спиртов типа бензилового спирта (SN 1).

Это объясняется стабильностью бензилового карбкатиона с участием бензольного кольца.
Спирты с β -расположением гидроксильной группы легко дегидратируются в арилолефины

Глава 15

