




Прежде чем передать файл для расчетов в Gaussian03 необходимо задать основные расчетные параметры. Минимальный набор в себя включает: тип расчета, метод расчета, квантово-механический базис, заряд молекулярной структуры, спиновая мультиплетность, название выходного файла. На контрольной панели задач редактора GaussView заходим в меню Calculate → Gaussian Calculation Setup, появляется окно Gaussian Calculation Setup (рис.2.1), содержащее следующие вкладки:
- Job Type;
- Method;
- Title;
- Link 0;
- General
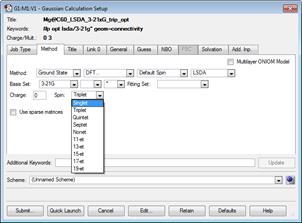
Рис.2.1 Окно Gaussian Calculation Setup
Во вкладке Job Type задается тип расчета:
- Energy – расчет энергии заданной молекулярной структуры;
- Optimization -запуск процесса оптимизации, заключающегося в расчете равновесных расстояний и конфигураций молекулярных структур, соответствующих минимальному значению полной энергии.
- Frequency- расчет вибрационного спектра;
- Opt+Frequency – выполнение процесса оптимизации с последующим расчетом вибрационного спектра;
- IRC – моделирование путей реакций;
- Scan – сканирование поверхности потенциальной энергии;
- Stability - переоптимизация волновой функции;
- NMR – расчет спектра ядерно-магнитного резонанса.
Примечание: Значения равновесных характеристик молекулярных структур, рассчитанные разными методами и базисами, отличаются друг от друга. Поэтому перед началом любого типа расчетов желательно провести процесс оптимизации несколькими методами и базисами, тогда вы сможете определить наиболее подходящий метод для расчетов вашего типа структур, например, сравнив длины связи с литературными данными.
Во вкладке Method пользователь задает расчетный метод, базис, спиновую мультиплетность, заряд молекулы. Выбирается метод расчета (HF – метод Хартри Фокка, DFT – метод теории функционала плотности, mechanics – молекулярная динамика, semi-empirical - полуэмпирические). Следующая опция позволяет устанавливать ограниченные restricted расчеты (на каждой молекулярной орбитали по два электрона с разным направлением спина) и неограниченные unrestricted (для каждого электрона своя молекулярная орбиталь и значение энергии). По умолчанию используется значение параметра Default Spin, т.е. при четном числе электронов в системе расчеты будут ограниченными, в случаи нечетного числа - неограниченными. В Basis Set задается, во-первых, основной базисный набор (подробнее см. пункт 2.1.3 данной лабораторной работы) и расширение набора за счет добавления поляризационных (*, **) или/и диффузных (+, ++) функций. Расчет ионизированных состояний молекулярных структур возможен за счет параметра Charge. По умолчанию используется значение «0», т.е. структура нейтральна. Устанавливая значение «+1» вы, тем самым, удаляете один электрон с внешней оболочки.
Параметр Spin позволяет задавать значение спиновой мультиплетности, величина которой определяется как 2S+1, где S- полный электронный спин системы. Если число электронов в системе четное, то, по умолчанию, GaussView использует значение S=0 (2 S +1=1), данное состояние называется синглетным. При нечетном числе электронов, по умолчанию, устанавливается низкоспиновое дублетное состояние S =1/2.
В свободном поле вкладки Title указываем название файла.
Максимальный объем выделяемый оперативной и памяти, место сохранения промежуточных результатов, количество используемых процессоров задается в Link 0. (см. лабораторную работу №1, пункт 1.1.1);
В General доступны дополнительные опции расчетов (критерий сходимости, использование симметрии и др.).

