




МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ
РОССИЙСКОЙ ФЕДЕРАЦИИ
Государственное общеобразовательное учреждение высшего профессионального образования
ВОРОНЕЖСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
НАНОТЕХНОЛОГИИ И НАНОМАТЕРИАЛЫ
ВЫЧИСЛИТЕЛЬНЫЙ ПРАКТИКУМ
Часть 1. КОМПЬЮТЕРНОЕ МОДЕЛИРОВАНИЕ ЭЛЕКТРОННОЙ СТРУКТУРЫ ФУЛЛЕРЕНА С60
Учебно-методическое пособие
Битюцкая Л.А., Тучин А.В., Бормонтов Е.В.
ББК 32.97
К
УДК 538.915:532:004
Составители:
Л.А. Битюцкая, А.В. Тучин, Е.Н. Бормонтов
Подписано в печать.11
Гарнитура «Times New Roman». Бумага офсетная.
Формат 60х84 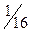 . Объем 3 п.л.
. Объем 3 п.л.
Тираж 200 экз. Заказ.
Компьютерное моделирование электронной структуры фуллерена С60: Вычислительный практикум / Сост. Л.А. Битюцкая, А.В. Тучин; Е.Н. Бормонтов.- Вып.1. – Воронежский государственный университет, 2011.- 96 с.; ил.24, прил. 4.- (Нанотехнологии и наноматериалы).
ISBN 978-5-91359-095-4
Учебно-методическое пособие “Компьютерное моделирование электронной структуры фуллерена С60” является первым выпуском цикла практических работ “нанотехнологии и наноматериалы” серии вычислительного практикума в программном комплексе Gaussian03 с использованием редактора GaussView. Выпуск включает четыре лабораторные работы, позволяющие познакомиться c современными методами вычислительной физики и химии: методом теории функционала плотности и методом Хартри-Фока, приобрести практические навыки численного моделирования электронной структуры, молекулярного строения и ИК-спектра самоорганизованных кластеров и молекул, таких как фуллерены. Учебное пособие предусматривает многоуровневые по сложности задания, включая творческие. Практикум предназначен для магистров по направлениям: “электроника и наноэлектроника”, “физика” и “химия, физика и механика материалов”, также может использоваться слушателями факультета повышения квалификации по направлению “нанотехнологии и наноматериалы” и студентами старших курсов и аспирантов при выполнении НИР.
ББК 32.97
УДК 538.915:532:004
ISBN 978-5-91359-095-4 © Воронежский государственный
университет, 2012
Оглавление
Введение. 6
Лабораторная работа №1 «Основы работы в редакторе GaussView пакета Gaussian03» 8
1.1 Теоретическая часть. 8
1.1.1 Программный комплекс Gaussian03. 8
1.1.2 Редактор GaussView.. 11
1.2 Практическая часть. 15
1.2.1 Построение молекул c использованием библиотек редактора GaussView 15
1.2.2. Примеры задания конфигурации молекул в редакторе GaussView 16
Вопросы.. 20
Лабораторная работа №2 «Численное моделирование электронной структуры молекул с использованием пакета Gaussian03». 21
2.1 Теоретическая часть. 21
2.1.1 Основные методы расчета молекулярных структур. 21
2.1.2 Неэмпирические методы расчета. 22
2.1.2.1 Метод Хартри-Фока. 22
2.1.2.2 Метод теории функционала плотности. 24
2.1.3 Основные квантово- механические базисы.. 27
2.2 Практическая часть. 30
2.2.1 Использование Gaussian Calculation Setup для установки параметров расчетов 30
2.2.2 Контрольный пример. Расчет характеристик молекулы кислорода 32
2.2.2.1 Визуализация электронной структуры атома кислорода. 32
2.2.2.2 Зависимость полной энергии двух атомов кислорода от расстояния между ними. 36
2.2.2.3 Расчет равновесного расстояния молекулы кислорода. 40
Вопросы.. 42
Лабораторная работа №3 «Молекулярное строение и электронная структура молекулы фуллерена С60». 44
3.1 Теоретическая часть. 44
3.1.1 Молекулярное строение фуллеренов C60 44
3.1.2 Получение фуллеренов. 46
3.1.3 Свойства фуллеренов. 49
3.1.4 Применение фуллеренов. 52
3.1.5 Фуллериты.. 58
3.1.6 Электронная структура фуллеренов С60 59
3.2 Практическая часть. 61
3.2.1 Электронная структура молекулы фуллерена С60 61
Вопросы.. 62
Лабораторная работа №4 «ИК- спектр изолированной молекулы фуллерена С60 и в присутствии растворителя». 64
4.1 Теоретическая часть. 64
4.1.1 ИК-спектроскопия как метод идентификации молекул. 64
4.1.2 Колебательный спектр двухатомной молекулы.. 66
4.1.3 Колебания многоатомных молекул. 70
4.1.2 ИК-спектр фуллерена С60 71
4.2 Практическая часть. 74
4.2.1 Расчет колебательного спектра молекулы воды в Gaussian. 74
4.2.2 ИК-спектр молекулы фуллерена С60 75
4.2.3 Учет эффектов сольватации при расчетах в Gaussian03. 76
4.2.4 ИК-спектр молекулы С60 в растворах. 77
Вопросы: 78
Литература. 79
Глоссарий терминов. 80
Приложение 1. Панель меню и панель команд редактора GaussView. 81
Приложение 2. Справочная информация о неорганических молекулах. 83
Приложение 3. Справочная информация об органических молекулах. 89
Приложение 4. Правила оформления лабораторных работ. 97
Введение
В 1996 году в Стокгольме вручена Нобелевская премия по химии Ричарду Смолли, Роберту Керлу и Гарольду Крото, ставшая венцом экспериментального открытия фуллеренов в 1985 году – нового класса молекул углерода. Лауреаты отмечают, что «открытие состояло в установлении того факта, что углерод один, без посторонней помощи образует молекулы в форме усеченного икосаэдра и более крупные геодезические клетки», при этом «углерод изначально, с момента возникновения Вселенной, одарен этой способностью к самопроизвольной сборке молекул фуллеренов». Особое место среди всех фуллеренов занимает молекула C60. Во-первых, благодаря высокой симметрии и стабильности, во-вторых, из-за технологичности получения и очистки. Возможность существования молекулы С60 замкнутой формы, напоминающей покрышку футбольного мяча, не раз обсуждалась исследователями в научной литературе. Советскими учеными: Бочваром, Гальперном и Станкевичем была показана стабильность молекулы C60 на основании численных расчетов еще за 10 лет до экспериментального открытия этой интереснейшей формы углерода. Многие уникальные оптические, электрофизические и химические фуллеренов, предсказанные с помощью методов квантовой химии, были также успешно подтверждены экспериментами.
История открытия и изучения фуллеренов показала, что совместное применение численных расчетов и эксперимента (например, ИК- и ЯМР- спектры) для изучения электронной структуры самоорганизованных углеродных нанокластеров является передовым направлением современной науки. Постоянное развитие мощности компьютеров привело к тому, что численное моделирование стало одним из самых важных инструментов исследований в физике, химии, биологии, материаловедении и др. Компьютерное моделирование структур и свойств веществ не только дополняет экспериментальные методы исследования, но и во многих случаях позволяет получать новые данные. Существенно сокращаются временные и материальные затраты на практические поиски новых соединений или внешних условий, при которых исследуемая молекула начинает обладать желаемыми свойствами. Применение вычислительных кластеров и специализированных программных комплексов широко используется ведущими исследовательскими университетами и предприятиями мира и является залогом их успешного развития.
На данный момент существует достаточное количество программных комплексов для расчета квантово-химических задач. Одним из самых распространенных и хорошо зарекомендовавших себя является пакет программ Gaussian03.
Цель вычислительного практикума по нанотехнологиям - изучение электронного строения и спектров инфракрасного поглощения фуллеренов С60 как самоорганизованных нанообъектов с использованием программного комплекса Gaussian03. В практикум входит 4 лабораторные работы:
1. «Основы работы в редакторе GaussView пакета Gaussian03»
2. «Численное моделирование электронной структуры молекул с использованием пакета Gaussian03»
3. «Молекулярное строение и электронная структура молекулы фуллерена С60»
4. «ИК- спектр изолированной молекулы фуллерена С60 и в присутствии растворителя»
Лабораторная работа №1 «Основы работы в редакторе GaussView пакета Gaussian03»
Цель работы: знакомство с программным комплексом Gaussian03, приобретение навыков работы в редакторе GaussView (задание геометрии расчетной структуры и др.).
Теоретическая часть
Программный комплекс Gaussian03
Для изучения электронной структуры как основных, так и возбужденных, часто короткоживущих, состояний молекул, применяется квантово-механическое моделирование в специальных программных комплексах. В случае правильно выбранного метода моделирования, данные, полученные в результате численных расчетов, хорошо согласуются с результатами экспериментов. Одним из самых распространенных программных пакетов является Gaussian. Программы серии Gaussian с момента своего возникновения (1970г.) отличались высокой степенью эффективности. В каждой новой версии Gaussian используются новейшие достижения в области квантовой химии. Руководитель группы разработчиков данного программного комплекса Джон Попл за развитие методов квантовой химии удостоен Нобелевской премии по химии 1998г.
Пакет может быть использован для изучения молекул при широком наборе условий, включая неустойчивые состояния молекул, которые трудно или невозможно наблюдать в экспериментальных условиях. Основные возможности пакета:
- Моделирование электронных структур молекул, кластеров, биологических соединений;
- Моделирование периодических систем, таких как полимеры и кристаллы, посредством использования периодических граничных условий;
- Моделирование широкого диапазона спектров и спектроскопических свойств молекул;
- Расчет энергий связей и путей реакций;
- Моделирование свойств молекул в растворах.
Gaussian состоит из набора программ - линков, решающих определенные задачи. Линки, выполняющие близкие функции, объединены в оверлеи. Линки имеют трех или четырехзначный номер. Последние две цифры обозначают номер линка в оверлеее.
Оверлей 0 – интерпретирует раздел директив входного файла;
Оверлей 1 – считывает исходные данные и следит за выполнением процессов оптимизации;
Оверлей 2 – определяет центр масс и симметрию молекулы;
Оверлей 3 – управляет выбором базисных функций и вычислением интегралов (одноцентровых, мультипольных, двухэлектронных и др.);
Оверлей 4 – формирует начальный набор молекулярных обиталей, выполняет расчеты с использованием полуэмперических меотодов и методов молекулярной механики.
Оверлей 5 – содержит линки, осуществляющие процедуры самосогласованного поля;
Оверлей 6 – производится анализ атомно-молекулярных систем на основе волновых функций, полученных в оверлее 5;
Оверлей 7 – расчет первых и вторых производных интегралов;
Оверлей 8 – выполняет преобразование атомных (одноцентровых) интегралов в молекулярные (многоцентровые);
Оверлей 9 – выполняет проверку стабильности волновых функций и ее переоптимизацию;
Оверлей 10 – выполняет процедуры самосогласования при использовании сложных методов, расчет спектров ядерно-магнитного резонанса;
Оверлей 11 – предназначен для вычисления производных одноэлектронных интегралов, матричных элементов от производных дипольного момента
Линк L9999 – выполняет окончание вычислений и формирует выходной файл.
Перед выполнением расчетов необходимо задать геометрию моделируемой структуры – положения, расстояния и углы между атомными ядрами. Пример входного файл комплекса Gaussian для расчета энергии молекулы воды представлен на рис.1.1.

Рис. 1.1.Входной файл комплекса Gaussian для расчета молекулы воды
В разделе директив указываются:
%mem – максимальное количество оперативной памяти, которое может использовать программа;
%chk – имя chek-point файла, в котором содержится информация о промежуточных вычислениях;
%nproc – число используемых процессоров;
# - данные о промежуточных расчетах выводятся в стандартной форме (#T – в сжатой, #P – в развернутой).
Далее следует описание расчетного метода (в данном случае LSDA), базиса (3-21G) и максимального количества выделяемой памяти на жестком диске (подробнее о методах расчетах и базисах вы узнаете в лабораторной работе №2).
В разделе спецификации расчетной структуры указывается элементный состав, координаты и тип связи между атомами. Кратность связей обозначается цифрами: 1.0 – одинарная, 1.5 – ароматическая, 2.0 – двойная и т.д. Запись «1 2 1.0 3 1.0» означает, что 1 и 2, 1 и 3 атомы связаны одинарными связями.
Для небольших молекул несложно задать пространственное положение атомов, однако, если необходимо произвести расчеты для больших структур, таких как фуллерен, то задача сильно усложняется. Значительное время тратится на построение молекул, а не на расчет их свойств, возрастает количество пользовательских ошибок. Что же делать, если необходимо рассчитать системы с числом атомов ~103? Для решения данной проблемы (и многих других) существует редактор GaussView.
Редактор GaussView
Графический редактор GaussView позволяет легко конструировать рассчитываемые молекулярные и кластерные структуры в удобном и наглядном для пользователя представлении. Библиотеки редактора GaussView содержат не только атомы, но и молекулы, радикалы, биологические молекулы, существует возможность создавать пользовательские структуры и пополнять ими библиотеку. Все это в сумме позволяет значительно ускорить процесс построения моделируемых структур.
Во-вторых, с использованием редактора GaussView задается используемый расчетный метод. Создание пользовательских схем расчетов (т.е. метод, базис, выделяемый объем памяти и др., используемые по умолчанию) позволяет сократить время на подготовку входного файла (подробнее о настройке параметров при выполнении расчетов будет рассмотрено в лабораторной работе №2). После построения расчетной структуры, установки метода расчета и дополнительных опций редактор генерирует входной Gaussian Job File в формате *.gjf (для операционной системы Windows) или *.com (для Linux). Запуск расчетов в Gaussian можно осуществлять с помощью редактора.
По окончании расчетов в комплексе Gaussian формируется файл, содержащий данные результатов расчетов в формате *.log или *.out в зависимости от операционной системы. Таким образом, третьей важной задачей редактора GaussView является перевод результатов расчетов в графическое наглядное представление, построение различных графиков. GaussView позволяет визуализировать:
- оптимизированную молекулярную структур, отвечающую минимуму энергии;
- молекулярные орбитали;
- поверхности и контуры электронной плотности;
- заряды атомов;
- анимацию вибрационных мод;
- ИК-, ЯМР- и Рамановские спектры;
Внешний вид редактора представлен на рисунке 1.2.
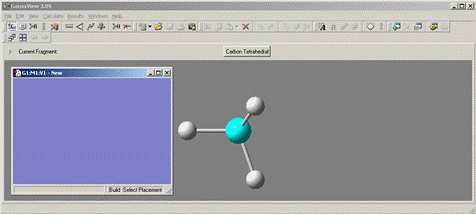
Рис. 1.2. Контрольная панель и рабочее окно редактора GaussView.
В состав панели меню на контрольной панели редактора входят:
- File: создание, открытие и сохранение структур, установка параметров редактора
- Edit: построение молекулярных структур
- View: управление настройками отображения
- Calculate: параметры расчетов и передача входных данных в Gaussian.
- Results: просмотр результатов расчетов, включая графики, спектры, поверхности и анимации;
- Windows: управление окнами GaussView.
Табл.1.1. Панель команд редактора GaussView

| |||

| библиотека элементов Element Fragment | 
| изменение длины и типа химических связей Modify Bond |

| библиотека циклических соединений Ring Fragment | 
| изменение величины валентных углов Modify Angle |

| библиотека радикалов R-Group Fragment | 
| изменение величины дигедральных углов Modify Dihedral |

| библиотека биомолекул Biological Fragment | 
| увеличение валентности Add Valence |

| база пользователя Custom Fragment | 
| удалить атом Delete Atom |
| |||

| численное задание координат Atom List Editor | 
| кристаллический редактор PBC Editor |

| дополнительные параметры координат Redundant Coordinate Editor | 
| редактор молекулярных орбиталей MO Editor |

| |||
| вырезать выделенное | 
| отменить последнее действие |

| копировать выделенное | 
| перерасчет длин связей |

| вставить из буфера обмена | 
| проверка геометрии на соответствие правилам |

| удалить выделенное | 
| определение точечной группы симметрии |
Табл.1.2 Использование мыши в редакторе GaussView
| функция | действие мышью в рабочем окне |
| выделение или вставка атома | нажать левую кнопку |
| вращение всех объектов | зажать левую кнопку |
| вращение выделенной молекулы | зажать Alt+ левая кнопка |
| вращение в плоскости рабочего окна | зажать Alt+ правая кнопка/ Ctrl+ (Left, Right) |
| перемещение | зажать Shift+ левая кнопка/ средняя кнопка/ Shift+(Left, Right, Up, Down) |
| перемещение выделенной молекулы | зажать Alt+ средняя кнопка |
| вызов контекстного меню | нажатие правой кнопки |
| приблизить/удалить | зажать Ctrl +левая кнопка/ правая кнопка/ Ctrl +(Up, Down) |
Практическая часть

