




Активований комплекс - групування частинок, що знаходяться в процесі взаємодії (тобто перерозподіл зв’язків). Перехідний енергетично нестійкий стан реагуючих речовин.
Активні частинки - реакційноздатні частинки (молекули, атоми, радикали), які володіють енергією, достатньою для переходу енергетичного бар’єру і утворення активованого комплексу.
Гетерогенні реакції - реакції між речовинами, що знаходяться в різних фазах. Проходять на поверхні розділу фаз.
Гомогенні реакції - реакції між речовинами в однофазному середовищі. Проходять по всьому об’єму системи.
Гетеролітичний розрив зв’язку - розрив хімічного зв’язку з переходом електронної пари зв’язку до більш електронегативного атома, в результаті чого утворюються йони.
Гомолітичний розрив зв’язку - розрив хімічного зв’язку при якому кожна частина молекули отримує по одному електрону електронної пари зв’язку. В результаті утворюються радикали.
Ініціювання ланцюгової реакції - перша стадія ланцюгової реакції, в результаті чого утворюються радикали.
Йонний механізм реакції - проходження реакції з проміжною участю йонів у елементарному акті реакції.
Істинна швидкість реакції - швидкість реакції в нескінченно малий проміжок часу.
Каталіз - збільшення швидкості реакції, що зумовлене наявністю в зоні реакції каталізатора.
Каталізатор - речовина, що прискорює реакцію, але не витрачається в процесі реакції.
Константа рівноваги - відношення добутку рівноважних концентрацій (активностей) продуктів оборотної реакції, взятих в степенях їх стехіометричних коефіцієнтів до аналогічного добутку для вихідних речовин цієї реакції.
Константа швидкості реакції - коефіцієнт пропорційності в кінетичному рівнянні закону діючих мас.
Молекулярність реакції - число частинок, що приймають участь у елементарному акті хімічної взаємодії.
Молекулярний механізм реакції - проходження реакції з проміжною участю молекул (атомів) у елементарному акті реакції.
Необоротна реакція - реакція, яка проходить лише в прямому напрямку в результаті якої утворюються продукти, що не взаємодіють між собою.
Оборотна реакція - реакція, що проходить при заданій температурі в двох протилежних напрямках з вимірюваними швидкостями.
Порядок реакції (окремий) - показник степеня при концентрації речовин в диференційному кінетичному рівнянні. Сумарний порядок реакції - сума порядків всіх реагентів.
Радикал - частинка, що має неспарений електрон і утворилась при гомолітичному розриві зв’язку.
Радикальний механізм реакції - проходження реакції з проміжною участю радикалів в елементарному акті реакції.
Швидкість гетерогенної реакції - кількість речовини, що реагує (утворюється) за одиницю часу на одиниці поверхні розділу фаз.
Швидкість гомогенної реакції - кількість речовини, що реагує (утворюється) за одиницю часу в одиниці реакційного об’єму.
Сорбція - загальна назва процесу, в якому проходить поглинання речовини одної фази речовиною іншої фази.
Середня швидкість реакції - зміна кількості речовини, що приймає участь в реакції за одиницю часу.
Хімічна кінетика - розділ хімії, що вивчає швидкість хімічних реакцій, залежність швидкості реакцій від різних факторів, а також механізм проходження реакції.
Хімічна рівновага - рівновага в хімічній системі, коли при незмінних зовнішніх умовах здійснюється рівність швидкостей прямих і обернених хімічних реакцій.
Енергія активації - мінімальна надлишкова (по відношенню до середньої) енергія реагуючих частинок, достатня для здійснення хімічної взаємодії.
Ефективне зіткнення - зіткнення реагуючих молекул, що приводить до утворення активованого комплексу.
Гетерогенна система - система, що складається з двох, або більшого числа фаз.
Гомогенна система - система, що складається із однієї фази.
Фаза - сукупність однорідних частин системи, що відділені від інших частин системи поверхнею розподілу.
Система (термодинамічна) - сукупність тіл (речовин), які фізично (уявно) відділені від навколишнього середовища.
Приклад 2. Що відбувається з хімічними зв’язками під час хімічних реакцій різних систем з точки зору хімічної кінетики?
Відповідь:
Хімічні реакції зводяться до розриву зв’язків у вихідних речовинах і виникненню нових зв’язків у продуктах реакції. При цьому загальне число атомів кожного елемента до та після реакції залишається постійним. Оскільки утворення зв’язків проходить з виділенням, а розрив - з поглинанням енергії, хімічні реакції супроводжуються енергетичним ефектом. Очевидно, якщо зв’язки, які розриваються у вихідних речовинах менш міцні, ніж ті, що утворюються в продуктах реакції, то енергія виділяється, і навпаки. Як правило, енергія виділяється та поглинається у формі теплоти.
Зі швидкістю хімічних реакцій зв’язані уявлення про перетворення речовин, а також економічну ефективність їх одержання в промислових масштабах. Вчення про швидкість хімічних реакцій та їх механізм називається хімічною кінетикою. Дослідження кінетики хімічних процесів має не тільки теоретичний, але й практичний інтерес. Необхідність врахування кінетичного фактору при розгляді хімічних реакцій можна побачити на прикладі водню та кисню. Незважаючи на те, що реакція:
2 H2(г) + O2(г) = 2 H2O(г)
може проходити самостійно, за звичайних умов водень та кисень між собою не реагують, а їх суміш може зберігатися будь-який час. При наявності каталізатора або при нагріванні до 7000С суміш реагує дуже швидко, а інколи навіть з вибухом.
Розглядаючи питання хімічної кінетики, треба розрізняти гомогенні та гетерогенні реакції. Гомогенною називається система, що складається з однієї фази, відповідно реакції, що в ній відбуваються - гомогенні.
Гетерогенна система - система, що складається з декількох фаз, і відповідні реакції - гетерогенні, тобто ті, що відбуваються в неоднорідному середовищі між речовинами, які перебувають у різних фазах (газ та рідина, кристал та рідина, тощо). Якщо гомогенні реакції відбуваються у всьому об’ємі системи, то перебіг гетерогенних реакцій можливий лише на поверхні розділу фаз.
Фаза - частина системи відділена від інших поверхнею розділу, при переході через яку властивості речовини змінюються.
Прикладами гомогенних реакцій є будь-які газові суміші (всі гази при не дуже великих тисках необмежено розчиняються), наприклад суміш азоту з киснем (повітря); розчин декількох речовин у одному розчиннику, наприклад розчин натрій хлориду, магній сульфату, азоту і кисню у воді. У кожному з цих випадків система складається лише з однієї фази: з газової чи рідкої. Гетерогенні системи - вода з льодом, насичений розчин з осадом, тверді частинки вугілля і сірки в атмосфері повітря. У всіх цих випадках в системі в наявності як мінімум дві фази.
Приклад 3. Що характеризує інтенсивність хімічного процесу?
Відповідь:
Швидкість хімічних реакцій характеризує інтенсивність хімічного процесу, тобто число елементарних актів взаємодії або розкладання за одиницю часу в одиниці об’єму (для гомогенних реакцій) або на одиницю поверхні поділу фаз (для гетерогенних реакцій).
Для гомогенних процесів, що відбуваються без зміни об’єму, швидкість хімічної реакції визначають як зміну концентрацій реагуючих речовин або продуктів реакції за одиницю часу. Зміна концентрацій дорівнює різниці між концентрацією С2, що відповідає моменту часу t2, та початковою концентрацією С1 в момент часу t1. Тоді середня швидкість реакції дорівнює:
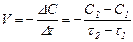
Чим менший проміжок часу Dt, тим менша зміна концентрацій Dс і тим ближче відношення Dс/Dt до істинної швидкості хімічної реакції. Точним математичним виразом істинної швидкості реакції є перша похідна від концентрації за часом, оскільки концентрації речовин у хімічному процесі змінюються безперервно.
|
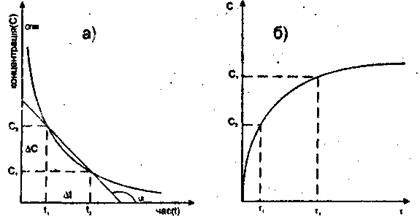
Рисунок 8.1 - Зміна концентрації С реагуючих речовин (а) та продуктів реакції (б).
Оскільки швидкість хімічних реакцій завжди додатна та характеризується зміною концентрацій реагуючих речовин (С2<С1), то величина Dс (dc) буде від’ємна і відношення 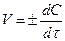 у формулах треба буде брати зі знаком „мінус”. Якщо швидкість реакції визначають за зміною концентрації одного з продуктів реакції, то величина Dс (dc) додатна і відношення у формулі треба брати зі знаком „плюс”. Одиниця вимірювання швидкості хімічної реакції - моль/м3´с (гомогенна), моль/м2´с (гетерогенна). Істинна швидкість реакції дорівнює тангенсу кута нахилу дотичної до кривої залежності концентрації від часу (див рис. 8.1) V = tg α. Значення швидкості, розраховані за зміною концентрацій вихідних речовин або продуктів реакції можуть бути різними, якщо всі коефіцієнти в рівнянні реакції не дорівнюють одиниці. Так, швидкість реакції: H2+I2 = 2HI виміряна за зміною концентрації НI, вдвоє більша за швидкість виміряну за зміною концентрації водню або йоду. Справді, концентрація HI більша за концентрацію водню в два рази, тому
у формулах треба буде брати зі знаком „мінус”. Якщо швидкість реакції визначають за зміною концентрації одного з продуктів реакції, то величина Dс (dc) додатна і відношення у формулі треба брати зі знаком „плюс”. Одиниця вимірювання швидкості хімічної реакції - моль/м3´с (гомогенна), моль/м2´с (гетерогенна). Істинна швидкість реакції дорівнює тангенсу кута нахилу дотичної до кривої залежності концентрації від часу (див рис. 8.1) V = tg α. Значення швидкості, розраховані за зміною концентрацій вихідних речовин або продуктів реакції можуть бути різними, якщо всі коефіцієнти в рівнянні реакції не дорівнюють одиниці. Так, швидкість реакції: H2+I2 = 2HI виміряна за зміною концентрації НI, вдвоє більша за швидкість виміряну за зміною концентрації водню або йоду. Справді, концентрація HI більша за концентрацію водню в два рази, тому 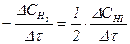 . Слід мати на увазі, що швидкість реакції може бути тільки додатною: якщо аналізувати швидкість реакції за зміною концентрації продуктів реакції, знак „мінус” у формулі не ставлять.
. Слід мати на увазі, що швидкість реакції може бути тільки додатною: якщо аналізувати швидкість реакції за зміною концентрації продуктів реакції, знак „мінус” у формулі не ставлять.
Коли говорять про зміну концентрації однієї з реагуючих речовин, не має значення, яку з них мають на увазі: всі вони зв’язані між собою рівнянням реакції, і по зміні концентрації однієї речовини можна судити про зміну концентрації інших речовин. Якщо, наприклад, вихідна концентрація однієї з реагуючих речовин складала 1 моль/л, а через 4 с від початку реакції вона стала 0,6 моль/л, тоді середня швидкість буде рівна (1 - 0,6)/4 = 0,1 моль/л´с.
Наведемо приклад гомо- та гетерогенних реакцій: зливання і перемішування розчинів сульфатної кислоти та натрій тіосульфату призводить до помутніння, що є наслідком появи сірки, спостерігається по всьому об’єму розчину:
H2SO4 + Na2S2O3 = Na2SO4 + H2O + SO2↑ + S↓
Розчинення металу в кислоті Fe + 2HCl = FeCl2 + H2 може проходити тільки на поверхні поділу фаз, що утворюють систему, тобто на поверхні металу, де проходить зіткнення реагуючих речовин. Швидкість хімічних реакцій залежить від природи реагуючих речовин, їх концентрації, температури, наявності каталізатора і деяких інших зовнішніх факторів.
Приклад 4. Вкажіть які фактори впливають на швидкість реакції. Сформулюйте закон діючих мас.
Відповідь:
Необхідною умовою перебігу хімічної реакції між двома речовинами є зіткнення їхніх молекул. Швидкість хімічної реакції залежить від числа таких зіткнень в одиниці об’єму. Ймовірність зіткнення взаємодіючих молекул для гомогенної реакції пропорційна концентрації реагуючих речовин. Звідсіля, на основі багатого експериментального матеріалу сформульовано основний закон хімічної кінетики, що встановлює залежність швидкості реакції від концентрації реагуючих речовин.
Швидкість хімічної реакції пропорційна добутку концентрацій реагуючих речовин у степенях, які дорівнюють коефіцієнтам, що стоять перед їх формулами у відповідному рівнянні реакції.
Ця закономірність має назву закону діючих мас і відкрита російським вченим М. М. Бекетовим та норвезькими вченими К. Гульдбергом та П. Вааге.
Для взаємодії двох молекул, наприклад водню та йоду, H2 + I2 = 2HI закон діючих мас (ЗДМ) у математичній формі має вигляд: 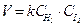
Для реакції: 2NO + O2 = 2NO2, 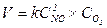
Для загальної реакції aA + bB = cC + dD швидкість дорівнює: V= kCAa´CBb = k [A]a´[B]b.
Величина k у рівнянні є коефіцієнтом пропорційності між швидкістю та концентрацією і називається константою швидкості реакції.
Неважко встановити фізичний зміст константи швидкості k: вона чисельно рівна швидкості реакції, якщо концентрації відповідних речовин рівні одиниці, або коли їх добуток рівний одиниці; константа швидкості при сталій температурі є величина стала і характеризує природу реагуючих речовин, тобто вона залежить від температури але не залежить від концентрації.
Рівняння, що зв’язує швидкість реакції з концентрацією реагуючих речовин називається кінетичним рівнянням реакції. Якщо дослідним шляхом визначене кінетичне рівняння реакції, тоді з його допомогою можна обчислити швидкості при інших концентраціях тих же речовин.
Основний закон хімічної кінетики не враховує реагуючі речовини, що знаходяться в твердому стані, оскільки їх концентрації постійні, і вони реагують лише на поверхні.
Так, наприклад, для реакції горіння вугілля С+О2 = СО2 ЗДМ запишеться: 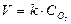 тобто швидкість реакції пропорційна тільки концентрації кисню. Постійними величинами в гетерогенному процесі є концентрація твердої фази та площа поверхні твердої фази (вугілля), добуток постійних величин позначений у рівнянні через k’.
тобто швидкість реакції пропорційна тільки концентрації кисню. Постійними величинами в гетерогенному процесі є концентрація твердої фази та площа поверхні твердої фази (вугілля), добуток постійних величин позначений у рівнянні через k’.
Для узагальнення матеріалу складемо дані рівняння швидкості реакцій:
| Хімічне рівняння | Cистема | Кінетичне рівняння |
| H2(г) + I2(г)= 2HI(г) | гомогенна | V = k[H2]´[I2],
де [H2] = 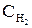
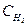 - концентрація, моль/л - концентрація, моль/л
|
| N2(г) + 3H2(г)= 2NH3 | гомогенна | V = k[N2]´[H2]3 |
| Fe(т) + 3Cl2(г) = = 2FeCl3(т) | гетерогенна | V = k[Cl2]3 |
| S(т) + O2(г) = SO2(г) | гетерогенна | V = k[O2] |
Сума показників степенів у рівнянні швидкості хімічної реакції (кінетичне рівняння) є важливою характеристикою механізму процесу і називається порядком хімічної реакції.
Розрізняють реакції першого порядку, наприклад:
N2O5 = 2NO2 + 1/2 O2 V = kC
реакції другого порядку, наприклад:
H2 + I2 = 2HI V = kC2; V = kC1C2
реакції третього порядку, наприклад:
2NO + O2 = 2NO2 V = kC3; V = kC12C2
Для характеристики механізму реакцій застосовують поняття молекулярності реакції (вивчають в наступних курсах хімії).
Підвищення температури реагуючих речовин внаслідок збільшення швидкості молекул зумовлює зростання загальної енергії системи і, відповідно, збільшення відносного вмісту активних молекул, що рівнозначно зростанню швидкості хімічної реакції.
Приклад 5. Яку роль в хімічних процесах відіграє енергія активації.
Відповідь:
Умовою елементарного акту взаємодії є зіткнення частинок реагуючих речовин. Проте не кожне зіткнення може спричинити хімічну взаємодію. Хімічна взаємодія передбачає перерозподіл електронної густини, утворення нових хімічних зв’язків і перегрупування атомів. Отже, крім зіткнення, енергія реагуючих частинок має бути більшою за енергію відштовхування (енергетичний бар’єр) між їхніми електронними оболонками. Внаслідок перерозподілу енергії частина молекул у системі завжди має певний надлишок енергії порівняно з середньою енергією молекул. Тому вони можуть подолати хімічний бар’єр та вступити в хімічну взаємодію. Такі реакційно здатні молекули дістали назву активних молекул. Різниця між середньою енергією системи та енергією, необхідною для перебігу реакції називається енергією активації реакції. Вона потрібна для подолання енергетичного бар’єру.
Наявність енергетичного бар’єру призводить до того, що багато реакцій, які цілком можуть проходити, самовільно не починаються. Наприклад, вугілля, деревина, нафта, які можуть горіти на повітрі за звичайних умов не займаються. Це пов’язано з великою енергією активації реакцій окиснення. Підвищення температури збільшує кількість активних молекул, і тому дедалі більше молекул кисню, вугілля деревини та нафти набувають необхідного запасу енергії для початку реакції. При певній температурі швидкість реакції досягає певної величини і починається реакція горіння. Отже, під час хімічного процесу перехід системи вихідних речовин з енергетичним станом Евих у енергетичний стан продуктів реакції Епр здійснюється через енергетичний бар’єр, який дорівнює енергії активації системи DЕакт. При цьому тепловий ефект реакції DН = Епр - Евих (DН - тепловий ефект реакції, ентальпія системи або її теплоємність). Позитивна величина зміни ентальпії DН відповідає зменшенню ентальпії, або виділенню теплоти системою.
Отже, залежність зміни швидкості реакції з зміною температури пояснює теорія активації. Згідно неї в хімічну взаємодію вступають тільки активні молекули, що володіють енергією, достатньою для проходження даної реакції. Один із способів активації - збільшення температури, що приводить до збільшення кількості активних молекул, які вступають у взаємодію.
Енергія, яку необхідно надати молекулам (частинкам) реагуючих речовин, щоб вони перетворилися в активні, називається енергією активації.
Її визначають дослідним шляхом, позначають символом Еа та виражають в кДж/моль. Так, наприклад, для реакції водню з йодом H2(г) + I2(г) = 2HI(г), Еа = 167.4 кДж/моль. Енергія активації Еа залежить від природи реагуючих речовин та служить характеристикою кожної реакції.
Вищезгадані теоретичні міркування можна пояснити на рис. 8.2.
|
|
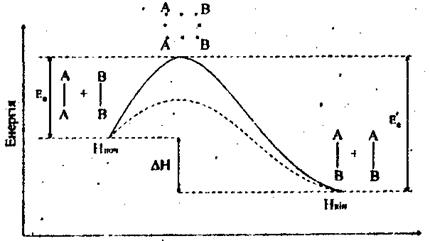

- активований комплекс
Еа - енергія активації прямої реакції, Еа’ - енергія активації зворотної реакції
Нпоч – енергія вихідного стану (вихідні речовини)
Нкін – енергія кінцевого стану (продукти реакції)
Рисунок 8.2 - Зміна енергії активації реагуючої системи.
На цьому рисунку зазначено проходження реакції А2 + В2 = 2АВ з точки зору теорії активації. Вісь ординат характеризує потенціальну енергію системи, вісь абсцис - хід реакції: вихідний стан → перехідний стан → кінцевий стан. Щоб реагуючі речовини А2 і В2 утворили продукт реакції АВ, вони повинні подолати енергетичний бар’єр С (див. рис. 8.2). На це затрачується енергія активації Еа, на значення якої збільшується енергія системи. При цьому в ході реакції з частинок реагуючих речовин утворюється нестійкий комплекс, що називається перехідним станом або активованим комплексом (в точці С), наступний розпад якого приводить до утворення кінцевого продукту АВ. Механізм реакції записується схемою:
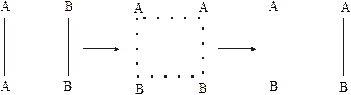
Якщо при розпаді активованого комплексу виділяється енергія більша, ніж необхідно для активації частинок, така реакція екзотермічна. Прикладом ендотермічної реакції може служити зворотній процес - утворення з речовини АВ речовин А2 і В2: 2АВ = А2 + В2. В цьому випадку процес проходить через також через утворення активованого комплексу, але енергія активації більша, ніж для першого процесу:
Еа’ = Еа + DН (DН - тепловий ефект реакції). Для проходження ендотермічної реакції необхідний підвід енергії ззовні.
Швидкість реакції безпосередньо залежить від значення енергії активації: якщо вона мала, то за певний проміжок часу проходження реакції енергетичний бар’єр долає більша кількість частинок, і швидкість реакції висока. Якщо енергія активації велика, тоді реакція проходить повільно.
При взаємодії йонів енергія активації мала, і йонні реакції проходять з великою швидкістю (практично зразу). Саме енергія активації затримує або робить неможливими багато реакцій, які можуть відбуватись самовільно з погляду термодинаміки.
Якби енергія активації була б рівна нулю (Еа = 0), то в природі відбувалося б безліч реакцій. Так, вугілля і нафта при контакті з повітрям загорілися б, азот повітря та вода утворили б розчин азотної кислоти, живі клітини зруйнувалися б внаслідок гідролізу.
Отже, існування багатьох молекул кристалічних речовин і навіть живих клітин можливе лише тому, що процеси їхнього перетворення та руйнування пов’язані з подоланням значного енергетичного бар’єру.
Приклад 6. Чомушвидкість реакції залежить від температури? Відповідь обґрунтуйте.
Відповідь:
Підвищення температури реагуючих речовин внаслідок збільшення швидкості молекул зумовлює зростання загальної енергії системи і, відповідно, збільшення відносного вмісту активних молекул, що рівнозначно зростанню швидкості хімічної реакції. Експериментально встановлено, що залежність швидкості хімічної реакції від температури можна виразити у вигляді емпіричного правила Вант-Гоффа: підвищення температури на кожні 10 градусів збільшує швидкість реакції в 2-4 рази.
Правило є наближеним та застосовується лише для орієнтовної оцінки впливу температури на швидкість реакції. Температура впливає на швидкість реакції, збільшуючи константу швидкості (тобто коефіцієнт пропорційності); крім того швидкість реакції залежить від енергії активації. Нагадаємо, що константа швидкості (k) залежить від природи реагуючих речовин та від температури, але не залежить від їх концентрації.
Математично правило Вант-Гофа виражається наступним чином:
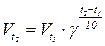
де Vt1, Vt2 - швидкості реакції відповідно при початковій (t1) та кінцевій (t2) температурах, а g - температурний коефіцієнт швидкості реакції, що показує в скільки разів збільшується швидкість реакції з підвищенням температури на 100С. (g = 2 - 4).
Вплив температури та енергії активації на швидкість реакції можна виразити за допомогою залежності константи швидкості реакції k від температури Т і енергії активації DЕакт:
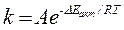
де А - множник Арреніуса, пропорційний числу зіткнень молекул. Якщо концентрації реагуючих речовин рівні 1 моль/л, то рівняння Арреніуса дає змогу виразити залежність швидкості реакції від температури:

Оскільки в рівнянні температура входить у показник степеня, то швидкість хімічних реакцій значною мірою залежить від температури.
Приклад 7. Оборотні та необоротні реакції. Хімічна рівновага. Принцип Ле - Шательє
Відповідь:
Всі хімічні реакції можна розбити на дві групи: оборотні та необоротні. Реакції, що проходять тільки в одному напрямку і завершуються повним перетворенням вихідних речовин в кінцеві продукти називаються необоротними.
Прикладом такої реакції може служити розклад калій хлорату (бертолетової солі) при нагріванні:
2KClO3 = 2KCl + 3O2
Вказана реакція проходить до кінця, тобто до тих пір, поки весь калій хлорат не перетвориться в калій хлорид та кисень. Необоротних реакцій не так багато, більшість оборотні. Оборотними називають такі реакції, які одночасно проходять у двох протилежних напрямках, тобто в прямому та зворотному.
Наприклад синтез аміаку з водню та азоту:
3H2 + N2 ↔ 2NH3, DН = -46,2 кДж/моль
У промисловості оборотні реакції, як правило, невигідні. Різними методами (зміною температури, тиску та концентрацій вихідних речовин) їх перетворюють в практично необоротні.
До оборотних реакцій належить наприклад взаємодія кисню з воднем:
2Н2 + О2 ↔ 2Н2О.
Справді, при температурі 800 - 15000С кисню з воднем утворюють воду, взаємодіючи досить бурхливо. При температурах 3000 - 40000С навпаки, вода розкладається з утворенням вихідних речовин. Взаємодія йоду з воднем
Н2 + I2 ↔ 2HI відбувається при температурі 300 - 4000С. При такій самій температурі можлива і зворотна реакція розкладу йодоводню. В наведених прикладах можна визначити швидкості прямої та зворотної реакції. Є також умови, за яких одночасно відбувається пряма та зворотна реакції. Проте відомі процеси, для яких визначити швидкість зворотної реакції неможливо, і тому можна лише говорити про оборотність процесу. Необоротними називають такі реакції при проходженні яких:
Продукт, який утворюється, виходить зі сфери реакції - випадає осад, виділяється газ, наприклад:
BaCl2 + H2SO4 = BaSO4 ¯ + 2HCl
Na2CO3 + 2HCl = 2NaCl + CO2 + H2O
Утворюються малодисоційовані сполуки, наприклад вода:
HCl + NaOH = NaCl + H2O
Реакція супроводиться великим виділенням енергії, наприклад горіння магнію:
Mg + 1/2 O2 = MgO, DН = -602 кДж/моль.
У рівняннях необоротних реакцій між лівою та правою частинами ставиться знак рівності, або стрілка.
Хімічна рівновага
Оборотні реакції не доходять до кінця, і закінчуються встановленням хімічної рівноваги. Наприклад, в реакції синтезу аміаку рівновага наступає тоді, коли в одиницю часу утворюється стільки ж молекул аміаку, скільки їх розпадається на азот та водень.
Значить: хімічну рівновагу можна визначити як такий стан системи реагуючих речовин, при якому швидкості прямої та оберненої реакції рівні між собою.
 В стані прямої та оберненої реакції не зупиняються. Така рівновага називається динамічною і в реагуючій суміші видимих змін не проходить: концентрації всіх речовин - як вихідних так і тих, які утворюються, залишаються виключно постійними. Концентрації реагуючих речовин, що встановлюються при хімічній рівновазі називаються рівноважними. Вони позначаються формулами реагуючих речовин, взятих у квадратні дужки, наприклад: [H2], [N2], [NH3], тоді як нерівноважні концентрації позначаються так:
В стані прямої та оберненої реакції не зупиняються. Така рівновага називається динамічною і в реагуючій суміші видимих змін не проходить: концентрації всіх речовин - як вихідних так і тих, які утворюються, залишаються виключно постійними. Концентрації реагуючих речовин, що встановлюються при хімічній рівновазі називаються рівноважними. Вони позначаються формулами реагуючих речовин, взятих у квадратні дужки, наприклад: [H2], [N2], [NH3], тоді як нерівноважні концентрації позначаються так:  (рис. 8.3).
(рис. 8.3).
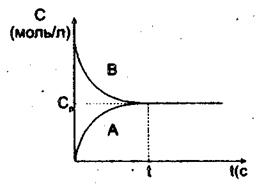
Ср – рівноважна концентрація (концентрація компонентів у стані рівноваги).
А – зменшення концентрації вихідних речовин.
В – збільшення концентрації продуктів реакції.
Рисунок 8.3 – Залежність концентрації від часу в стані хімічної рівноваги.
Кількісною характеристикою хімічної рівноваги служить величина, яка називається константою хімічної рівноваги. Розглянемо її на прикладі:
H2 + I2 ↔ 2HI.
Згідно закону діючих мас швидкості прямої та зворотної реакцій виражаються рівняннями:
V1 = k1[H2]´[I2]; V2 = k2[HI]2.
При рівновазі швидкості прямої та зворотної реакцій рівні, звідки:
k1[H2][I2] = k2[HI]2, або k1/k2 = [HI]2/[H2][I2]
Відношення констант швидкості прямої та зворотної реакцій також є константа і називається константою рівноваги даної реакції (K): k1/k2 = K.
У кінцевому плані: k = [HI]2/[H2][I2].
У правій частині цього рівняння знаходяться ті концентрації взаємодіючих речовин, які встановлюються при рівновазі - рівноважні концентрації. Ліва частина рівняння є величиною постійною при постійній температурі. Можна сказати, що у загальному випадку для оборотної реакції:
Aa + bB + … ↔ p  P + qQ + … константа виражається рівнянням:
P + qQ + … константа виражається рівнянням:
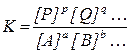
Великі букви позначають формули речовин, а маленькі - коефіцієнти у рівняннях реакцій.
Таким чином, при постійній температурі константа рівноваги оборотної реакції являє собою постійну величину, що показує співвідношення між концентраціями продуктів реакції (чисельник) та вихідних речовин (знаменник), що встановлюється при рівновазі.
У випадку гетерогенних реакцій у вираз константи рівноваги вносять концентрації лише тих речовин, які знаходяться в твердій або рідкій фазі.
Виходячи з виразу константи рівноваги, незалежно від початкових концентрацій, рівновага в системі встановлюється завжди при тому самому співвідношенні концентрацій продуктів реакції та реагуючих речовин. Це співвідношення не залежить від того, які речовини беруть за вихідні, наприклад: H2 + I2 або HI.
Отже, константа рівноваги залежить не від концентрації речовин, а від їх природи та температури реакції. У вираз константи хімічної рівноваги, як і у вираз константи швидкості, концентрації входять у степенях, які рівні стехіометричним коефіцієнтам у рівнянні реакції, воно є одним із виразів закону діючих мас.
На стан хімічної рівноваги впливає концентрація реагуючих речовин, температура, а для газоподібних - і тиск. При зміні одного з цих параметрів рівновага порушується, і концентрація всіх реагуючих речовин змінюється до тих пір, поки не встановиться нова рівновага, але вже при інших значеннях рівноважних концентрацій. Подібний перехід реакційної системи від одного стану рівноваги до другого називається зміщенням (зсувом) хімічної рівноваги. Якщо при зміні умов збільшується концентрація кінцевих речовин, тоді кажуть про зміщення рівноваги в сторону продуктів реакції. Якщо ж збільшується концентрація вихідних речовин, рівновага зміщується в сторону їх утворення.
Приклад 8. Сформулюйте принцип Ле-Шательє. Як впливають зовнішні фактори на хімічну рівновагу?
Відповідь:
Стан хімічної рівноваги за сталих умов може зберігатися будь-який час. Проте при зміні умов рівноваги (температури, тиску, концентрації) стан рівноваги порушується. Зміна зовнішніх факторів по різному впливає на швидкості прямої та зворотної реакцій, тобто швидкість однієї буде більша, ніж другої. Внаслідок цього хімічна рівновага зміститься в той чи інший бік. Через деякий час в системі знову встановиться рівновага, але вже за інших умов.
Характер зміни рівноваги залежно від зовнішніх факторів можна визначити за принципом Ле-Шательє. Цей принцип, відкритий у 1882 р. французьким вченим
Ле-Шательє, формулюється так: якщо на систему, що перебуває в рівновазі, подіяти зовнішнім фактором (зміною концентрації, температури, тиску), то рівновага зміститься у напрямку процесу, який зменшить цю дію.
Розглянемо принцип Ле-Шательє на прикладі рівноважної системи
3H2 + N2 ↔ 2NH3, DН = -42,6 кДж.
Якщо до суміші речовин, які перебувають у стані рівноваги, додати певну кількість водню або азоту, то швидкість прямої реакції збільшиться. Збільшення швидкості прямої реакції призведе до зменшення кількості реагуючих речовин (N2 та H2) і збільшення концентрації NH3. Система, з одного боку, зреагувала на зовнішній вплив. З другого - збільшення концентрації NH3 призведе до збільшення швидкості зворотної реакції, яке відбуватиметься доти, поки швидкість зворотної реакції не зрівняється із швидкістю прямої реакції, тобто в системі знову настає стан рівноваги, але вже за нових умов, а саме, при вищих швидкостях реакцій.
Вплив тиску на стан рівноваги можна проаналізувати, розглянувши зміну об’єму речовин в реакції синтезу аміаку. З рівняння реакції видно, що з чотирьох молекул реагуючих речовин (3H2 + N2) утворюється дві молекули NH3, тобто синтез аміаку відбувається зі зменшенням об’єму. Підвищення тиску для газоподібних систем зумовлює пропорційне зменшення об’єму, що відповідає збільшенню концентрацій речовин. Оскільки відносне збільшення концентрацій реагуючих речовин у реакції синтезу аміаку перевершуватиме збільшення концентрацій продуктів реакції, то рівновага зміститься в бік реакції, яка призводить до зменшення об’єму системи.
У реакціях, що відбуваються без зміни об’єму або в яких не беруть участі газоподібні речовини, зміна тиску не викликає зміщення рівноваги.
Підвищення температури зазначеної системи спричиняє зміщення рівноваги в бік зворотної реакції, оскільки розклад аміаку призводить до поглинання теплоти. Каталізатор не впливає на стан рівноваги, однаково прискорюючи як пряму, так і зворотну реакції. Тобто каталізатор прискорює встановлення рівноваги, не впливаючи на рівноважні концентрації речовин.
На основі аналізу зазначених прикладів можна сформулювати ряд загальних положень, які випливають із принципу Ле-Шательє, а саме: при підвищенні температури рівновага системи, що перебуває у стані рівноваги, зміщується у напрямку ендотермічної реакції, а при зниженні - у напрямку екзотермічної.
Підвищення тиску призводить до зміщення рівноваги в бік утворення тих речовин, які займають менший об’єм. Якщо об’єм системи не змінюється, то тиск невпливатиме на стан рівноваги.
Принцип Ле-Шательє справедливий не тільки для хімічних процесів, він має загальнонаукове значення і поширюється на всі процеси, що перебувають в стані динамічної рівноваги.
Принцип Ле-Шательє має велике практичне значення, особливо для хімічної промисловості. Наприклад, при синтезі NH3 підвищення температури зменшує вихід аміаку, тобто для підвищення виходу його реакцію треба проводити при низьких температурах. Проте стан рівноваги при цьому завдяки дуже малій швидкості встановлюється за такий великий проміжок часу, що практичне використання реакції буде неможливим. Тому синтез NH3 проводять при високій температурі (450-6000С), а для прискорення рівноваги застосовують каталізатор. В той же час при атмосферному тиску вихід аміаку становить всього 1%. Промислове використання реакції синтезу аміаку стало можливим лише при застосуванні високого тиску, який у 300-1000 разів перевищує атмосферний, зміщуючи рівновагу в бік підвищення вмісту NH3 до 30%. При такому виході процес синтезу аміаку стає придатним для промислового використання.
Приклад 9. Записати вираз рівняння швидкості для прямої реакції: а) N2(г) + 3H2/г/ = 2NH3/г/;
б ) CaCO3/к/ → CaO/к/ + CO2/г/
Відповідь:
а) V = k[N2]´[H2]3
б) оскільки карбонат кальцію тверда речовина і концентрація його в процесі реакції не змінюється, вираз рівняння швидкості матиме вигляд: V = k, тобто швидкість реакції за певної температури стала.
Приклад 10. Як зміниться швидкість реакції
2СО(г) + О2(г) = 2СО2(г), якщо об’єм, у якому відбувається реакція збільшити в два рази?
Відповідь:
Згідно закону діючих мас швидкість V до зміни об’єму рівна:
V = k[CO]2´[O2].
Якщо збільшити об’єм у два рази, то концентрація кожної з реагуючих речовин зменшиться у два рази. При нових концентраціях швидкість прямої реакції (V’) буде:
V’ = k[1/2CO]2´[1/2O2] = 1/8k[CO]2´[O2].
Значить, швидкість прямої реакції зменшиться у 8 разів.
Приклад 11. Як зміниться швидкість реакції
Fe2O3(к) + 3H2(г) =2Fe(к) + 3H2O(г), якщо початковий тиск в системі збільшити в два рази?
Відповідь:
До зміни тиску в системі швидкість реакції V = k[H2]3 (від концентрації твердої речовини швидкісь реакції не залежить). При збільшенні тиску у три рази концентрація реагуючих речовин також збільшиться у три рази. При нових концентраціях швидкісь реакції V’ = k[3H2]3 = 27k[H2]3. Швидкість реакції зросте в 27 разів.
Приклад 12. Як зміниться швидкість прямої реакції
2СО(г) + О2(г) = 2СО2(г), якщо тиск збільшити в три рази?
Відповідь:
Припустимо, що в початковий момент до підвищення тиску концентрації реагуючих речовин наступні: [СО] = а, [О2] = b. Швидкість реакції до підвищення тиску визначаємо згідно закону діючих мас:
V = k´[CO]2´[O2] = ka2b. Згідно рівняння Менделєєва-Клапейрона підвищення тиску в три рази приводить до підвищення концентрації кожного компоненту в три рази.
Таким чином, після підвищення тиску концентрації реагуючих речовин стануть рівними: [СО] = 3а, [О2] = 3b.
Швидкість реакції після підвищення тиску
V’ = k[CO]´[O2] = k(3a)23b = 27a2b.
Отже, при підвищенні тиску в три рази швидкість даної реакції збільшиться в 27 раз.
Приклад 13. Користуючись рівнянням Вант - Гоффа розрахувати за який час закінчиться хімічна реакція при 1000С, якщо при 00С вона закінчилася за 10 хв. Температурний коефіцієнт реакції рівний 3.
Відповідь:
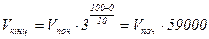 . Зі збільшенням температури на1000С хімічна реакція прискориться в 59000 разів. Якщо при 00С вона закінчилася за 10 хв. то при 1000С вона закінчиться за 600/59000 = 0,01 с.
. Зі збільшенням температури на1000С хімічна реакція прискориться в 59000 разів. Якщо при 00С вона закінчилася за 10 хв. то при 1000С вона закінчиться за 600/59000 = 0,01 с.
Приклад 14. У скільки разів збільшиться швидкість реакції при збільшенні температури з 50 до 900С? Температурний коефіцієнт реакції дорівнює 2,5.
Відповідь:
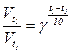 ;
; 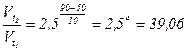
Приклад 15. Розрахувати у скільки разів збільшиться швидкість хімічної реакції, що протікає в газовій фазі, за умови збільшення температури від 30 до 700С. Температурний коефіцієнт реакції рівний 2.
Відповідь:
Залежність швидкості реакції визначається за правилом Вант-Гоффа:
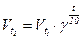 ;
; 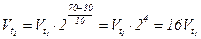 .
.
Отже швидкість хімічної реакції збільшиться в 16 разів.
Приклад 16. Як зміниться швидкість реакції у газовій фазі при зменшенні температури на 300С, якщо температурний коефіцієнт реакції рівний 3.
Відповідь:
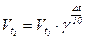 ;
;  .
.
Отже швидкість хімічної реакції зменшиться в 27 разів.
Приклад 17. Реакція при температурі 500С відбувається за
2 хв. 15 с. за який час закінчиться ця реакція при 700С, якщо в даному температурному інтервалі температурний коефіцієнт реакції рівний 3.
| Дано: t1 = 50 0C, t2 = 70 0C, Dt1 = 135 c, g = 3 |
| Dt2 =? |
Відповідь:
1) За формулою 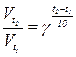 обчислюємо, у скільки разів збільшиться швидкість реакції при підвищенні температури від 500С до 700С:
обчислюємо, у скільки разів збільшиться швидкість реакції при підвищенні температури від 500С до 700С:
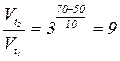
2) Відповідно до визначення швидкості 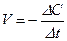 , де с(А) – молярна концентрація – кількість речовини, що міститься в одиниці об’єму, моль/м3с.
, де с(А) – молярна концентрація – кількість речовини, що міститься в одиниці об’єму, моль/м3с.
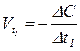 ;
; 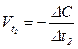 .
.
Оскільки при обох температурах t1 та t2 однакова, маємо:
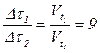
3) Визначаємо час, за який відбувається реакція при температурі 700С:
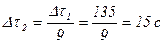 .
.
Приклад 18. Константа рівноваги гомогенної системи CO(г) + H2O(г) = CO2(г) + H2(г) при 8500С дорівнює 1. Розрахувати рівноважні концентрації всіх речовин, якщо відомо, що вихідні концентрації: [CO]вих. = 3 моль/л,
[H2O]вих. = 2 моль/л.
Відповідь:
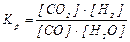 .
.
В умові задачі дано вихідні концентрації, а у вираз константи рівноваги входять тільки рівноважні концентрації всіх речовин. Припустимо, що в момент рівноваги [CO2]рівн. = x моль/л. Число моль водню в момент рівноваги також буде х моль/л. Тоді [CO] буде (3-х) моль/л, а [H2O] буде (2-х) моль/л. Підставивши ці значення концентрацій в рівняння константи рівноваги одержимо: 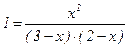 ; x2 = 6 - 5x + x2; 5x = 6; x = 1,2 моль/л.
; x2 = 6 - 5x + x2; 5x = 6; x = 1,2 моль/л.
Таким чином концентрації реагуючих речовин будуть:
[CO2] = 1,2 моль/л
[H2] = 1,2 моль/л
[CO] = 1,8 моль/л
[H2O] = 0,8 моль/л.
Приклад 19. Реакція протікає за рівнянням: N2 + H2 = 2NH3. Концентрації реагуючих речовин:
[N2] = 0,8 моль/л
[H2] = 1,5 моль/л
[NH3] = 0,1 моль/л. Обчислити концентрацію водню та аміаку в момент, коли [N2] = 0,5 моль/л.
Відповідь:
Концентрація азоту зменшилася на 0,8 - 0,5 = 0,3 моль/л. З рівняння видно, що з одного моля N2 утворюється
2 молі NH3.
З 1 моля N2 утворюється 2 молі NH3
З 0,3 моля N2 утворюється х моль NH3.
х = 0,6 моль/л.
Значить [NH3] = 0,1 + 0,6 = 0,7 моль/л.
Якщо прореагує 1 моль N2 то водню прореагує 3 моль
Якщо прореагує 0,3 моль N2 то водню прореагує х моль.
х = 0,9 моль.
Концентрація водню зменшиться на 0,9 моль/л. Значить H2 залишиться 1,5 - 0,9 = = 0,6 моль/л.
Приклад 20. Визначити початкову швидкість реакції з константою швидкості прямої реакції k в розчині, одержаному змішуванням 2 л 0,6М розчину СН3СООН і 3 л 1М розчину NH4OH.
Відповідь:
Рівняння хімічної реакції наступне:
NH4OH + CH3COOH ↔ NH4CH3COO + H2O.
Згідно з законом діючих мас вираз швидкості реакції
Vпр = k[NH4OH]´[CH3COOH].
Для визначення швидкості реакції необхідно знати величину концентрації речовин NH4OH та CH3COOH в момент їх змішування. Для цього визначимо об’єм суміші двох розчинів:
Vзаг = V1 + V2 = 2 + 3 = 5 л.
Знаючи, що у вихідному розчині оцтової кислоти міститься 0,6´2 = 1,2 моль, знаходимо її концентрацію в суміші:
[CH3COOH ] = 1,2моль/5л = 0,24 моль/л.
У вихідному розчині гідроксиду амонію міститься 1´3 = 3 моль, значить концентрація його в суміші:
[NH4OH] = 3моль/5л = 0,6 моль/л.
Визначаємо початкову швидкість реакції:
Vпр = k[NH4OH]´[CH3COOH] = k´0,24´0,6 = 0,144k.
Приклад 21. Поясність, до якого типу належать перелічені нижче хімічні реакції, і поясніть їх суть: а) взаємодія розчину ферум (ІІІ) хлориду з натрій гідроксидом; б) розкладання калій хлорату при нагрівання; в) окиснення сульфур (IV) оксиду; г) розкладання кальцій карбонату при нагріванні; д) взаємодія алюмінію з сіркою. Складіть рівняння цих реакцій.
Відповідь:
а) 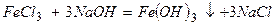 - необоротна обмінна реакція;
- необоротна обмінна реакція;
б) 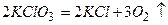 - необоротна окисно-відновна реакція;
- необоротна окисно-відновна реакція;
в) 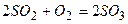 - оборотна окисно-відновна реакція;
- оборотна окисно-відновна реакція;
г) 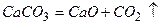 - необоротна реакція розкладу;
- необоротна реакція розкладу;
д) 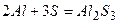 - необоротна реакція приєднання.
- необоротна реакція приєднання.
Приклад 22. Які умови впливають на швидкість хімічних реакцій? Наведіть для кожної умови один-два додаткових приклади, складіть рівняння реакцій і обґрунтуйте їх проходження.
Відповідь:
1) Швидкість хімічної реакції залежить від природи реагуючих речовин. Наприклад, галогени по-різному взаємодіють з воднем. Взаємодія фтору з воднем супроводжується вибухом при кімнатній температурі: 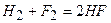 , а йод реагує із воднем тільки при сильному нагріванні:
, а йод реагує із воднем тільки при сильному нагріванні: 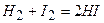 .
.
2) Для розчинів і газів швидкість хімічної реакції залежить від концентрації реагуючих речовин. Наприклад, швидкість реакції азоту з воднем: 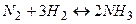 збільшується з підвищенням тиску, оскільки концентрація газів прямо пропорційна тиску.
збільшується з підвищенням тиску, оскільки концентрація газів прямо пропорційна тиску.
3) Для речовин з твердим агрегатним станом швидкість реакції залежить від поверхні реагуючих речовин. Подрібнений цинк скоріше взаємодіє із соляною кислотою:
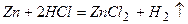
4) При підвищенні температури на кожні 10°С швидкість реакції збільшиться в 2 – 4 рази. Так, при кімнатній температурі залізо не реагує із концентрованою сірчаною кислотою, а при нагріванні розчиняється в ній:
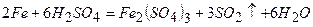
5) Швидкість хімічної реакцій залежить від присутності каталізаторів. Наприклад, залізо не реагує із сухим хлором, але швидко окислюється хлором в присутності вологи:

Приклад 23. Амоніак у промисловості одержують з водню та азоту. Нехай початкову концентрацію водню
[H2]n взяли 1,2 моль/л, азоту [N2]n=0,3 моль/л. Через деякий час концентрація водню знизилася до 0,6 моль/л. Якою на кінець терміну була концентрація азоту [N2]p?
Відповідь:
Дано:
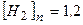 моль/л моль/л
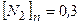 моль/л моль/л
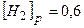 моль/л моль/л
| Розв’язок
1) Записуємо рівняння реакцій синтезу амоніаку 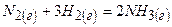
|
Знайти:
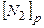 -? -?
| 2) Обчислюємо зменшення концентрації водню внаслідок реакції з азотом: |
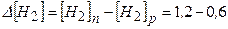 моль/л=0,6 моль/л
моль/л=0,6 моль/л
3) На основі рівняння реакції визначають зменшення концентрації азоту
3,0 моль 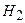 - 1 моль
- 1 моль 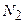
0,6 моль – х
х=0,2 моль
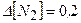 моль/л
моль/л
4) Рівноважна концентрація N2 дорівнює початковій мінус знайдене зменшення концентрації
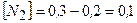 моль/л
моль/л
Відповідь: 0,1 моль/л.
Приклад 24. Як зміниться швидкість реакції 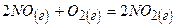 при зменшенні об’єму реагуючої суміші в 4 рази?
при зменшенні об’єму реагуючої суміші в 4 рази?
Дано:
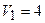 об’єми об’єми
 об’єм об’єм
| Розв’язок:
1. Відзначимо, що зменшення об’єму системи пропорційне збільшенню концентрації реагуючих речовин в ній. Тобто, якщо 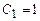 , то , то 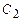 є в 4 рази більшою. є в 4 рази більшою.
|
Знайти:
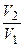 -? -?
|
2. Записуємо вирази для початкової і кінцевої швидкостей реакції
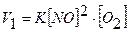
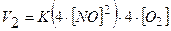
3. Обчислюємо відношення цих швидкостей
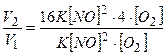
Відповідь: швидкість зросте в 64 рази.
Приклад 25. Визначити вихідні концентрації CO і H2O, коли відомо, що концентрації речовин в момент встановлення рівноваги
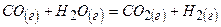
дорівнюють
[CO]p= 0,4моль/л, [H2O]p=0,3моль/л, [CO2]p=1,2моль/л
Дано:
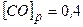 моль/л моль/л
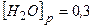 моль/л моль/л
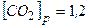 моль/л моль/л
| Розв’язок:
1. Знаходимо кількості речовин, що вступили в реакцію в 1л реакційної суміші
а) на 1 моль СO2 витрачається 1 моль 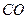 на 1,2 моль СO2 - ν моль CO.
на 1,2 моль СO2 - ν моль CO.
|
Знайти:
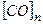 і і 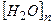
|
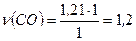 моль
моль
б) на 1 моль СO2 витрачається 1 моль H2O
на 1,2 моль СO2 - ν моль H2O.
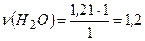 моль H2O
моль H2O
2. Вихідні концентрації речовин дорівнюють:
а) 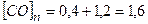 моль/л
моль/л
б) 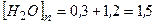 моль/л
моль/л
Відповідь: 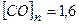 моль/л;
моль/л; 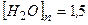 моль/л.
моль/л.
Приклад 26. Рівноважні концентрації речовин у реакції 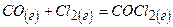 дорівнюють:
дорівнюють: 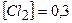 моль/л;
моль/л; 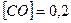 моль/л;
моль/л; 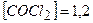 моль/л. Визначити константу рівноваги.
моль/л. Визначити константу рівноваги.
Відповідь:
1. Записуємо вираз для константи рівноваги реакції:
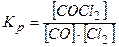
2. Обчислюємо константу:
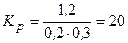
Відповідь: 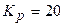 .
.
Приклад 27. У скільки разів зросте швидкість хімічної реакції, якщо температуру газової суміші підвищити від 40°С до 80°С. Температурний коефіцієнт реакції дорівнює 4.
Відповідь:
Дано:
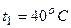
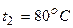

| Розв’язок:
|
| Знайти:
-?
|
 .
.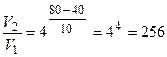
Відповідь: у 256 разів.
Приклад 28. Як вплине на рівноважний стан реакції 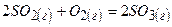
1) підвищення тиску
2) зменшення концентрації сульфур (VI) оксиду?
Відповідь:
Під час проходження прямої реакції зменшується число молекул газоподібних речовин:
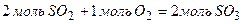
Згідно з принципом Ле Шательє внаслідок підвищення тиску рівновага в системі зміститься в бік зменшення числа молекул газів, тобто у бік зниження тиску – утворення газу SO3. Зменшення концентрації SO3 (виведення продукту реакції із системи) призведе до зміщення рівноваги у бік утворення 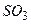 .
.
Приклад 29. Дано рівняння реакцій:
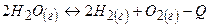
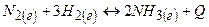
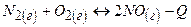
2SO2(г) + 2Н2О(г) ↔ 3 О2(г) + 2Н2S(г) - Q
В якій бік зміститься рівновага при підвищенні:
а) температури; б) тиску?
Відповідь:
При підвищенні температури рівновага зміщається в сторону ендотермічної реакції  , а при підвищенні тиску – в сторону зменшення загального числа молекул газів.
, а при підвищенні тиску – в сторону зменшення загального числа молекул газів.
1)
При підвищенні температури рівновага зміститься вправо, а при підвищенні тиску – вліво.
2)
При підвищенні температури рівновага зміститься вліво, а при підвищенні тиску – вправо.
3)
При підвищенні температури рівновага зміститься вправо, а при підвищенні тиску не зміниться, оскільки загальне число молекул газу в реакції не зміниться.
4) 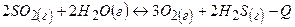
При підвищенні температури рівновага зміститься вправо, а при підвищенні тиску – вліво.
Приклад 30. Яку роль в хімічних процесах відіграють каталізатори?
Відповідь:
Збільшувати швидкість реакції можна за допомогою каталізаторів. Застосовувати каталізатори вигідніше, ніж підвищувати температуру, тим більше, що підвищувати її не завжди можливо. Каталізаторами називаються речовини, які змінюють швидкість хімічних реакцій. Хімічні реакції, які проходять за участю каталізаторів називаються каталітичними.
Сам каталізатор в процесі реакції кількісно та якісно не змінюється (не витрачається та в продукт реакції не входить). Одні каталізатори прискорюють реакцію - позитивний каталіз, інші сповільнюють - інгібітори реакції. Прикладом позитивного каталізу є одержання сульфатної кислоти, окиснення аміаку в азотну кислоту за допомогою платинового каталізатора. Прикладом уповільнення швидкості хімічного процесу є зменшення швидкості розкладу пероксиду водню в присутності невеликої кількості сульфатної кислоти.
Число каталізаторів досить велике, а їх каталітична активність надзвичайно різноманітна. Вона визначається зміною швидкості реакції під впливом каталізатора. Механізм дії каталізаторів різний. Найпоширенішою формою дії каталізатора є утворення проміжних сполук з реагуючими речовинами. Зміна швидкості реакції при цьому пояснюється зменшенням енергії активації. Розрізняють два види каталізу: гомогенний (однорідний) та гетерогенний (неоднорідний) каталіз. При гомогенному каталізі речовини, що реагують утворюють однофазну систему - газоподібну або рідку, між каталізатором та речовинами, що взаємодіють відсутня поверхня поділу. Наприклад каталітичний розклад пероксиду водню в присутності розчину солей (рідка фаза), окиснення СО в газоподібній фазі за наявності пари води як каталізатора (газова фаза). Добування сірчаної кислоти баштовим методом засноване на гомогенній каталітичній реакції
H2SO3 + NO2 = h2so4 + no, яка може відбуватися в газоподібній фазі.
При гетерогенному каталізі реагуючі речовини та каталізатор утворюють систему з різних фаз. В цьому випадку між каталізатором і речовинами, які взаємодіють є поверхня розділу, і реакція проходить на цій поверхні, тобто каталізатор утворює самостійну фазу (як правило тверду), де проходять каталітичні процеси. Прикладом гетерогенного процесу є окиснення SO2 до SO3 на поверхні V2O5, окиснення NH3 в присутності платини (газова/тверда фаза), розклад Н2О2 в присутності манган (IV) оксиду (рідка/тверда фаза).
Активність каталізатора залежить від властивостей його поверхні (розміру, хімічного складу, б

