




Второе начало термодинамики имеет особый статус не только фундаментального закона физики, но и универсального принципа естествознания в целом.
Второе начало дает информацию о направлении процессов, которые могут происходить в действительности. Оно, совместно с первым началом, позволяет установить множество точных количественных соотношений между различными макроскопическими параметрами тел в состоянии термодинамического равновесия. Это оказывается возможным благодаря определению такой величины как энтропия через теплоту. Энтропия (греч. – поворот, превращение) служит мерой преобразования или эволюции системы.
Существует несколько десятков различных формулировок второго начала. В большинстве из них термин «энтропия» не используется (см. формулировки Кельвина, Клаузиуса и Оствальда).
| Второе начало термодинамики | ||
| Формулировка Кельвина: Невозможен циклический процесс, единственным результатом которого является производство работы А за счет уменьшения внутренней энергии U только одного теплового резервуара (рис.13) | Формулировка Клаузиуса: Невозможен циклический процесс, единственным результатом которого является передача теплоты от менее нагретого тела к более нагретому (рис.14) | Формулировка Оствальда: Невозможно построить «вечный двигатель второго рода». |
Такие процессы невозможны. Они запрещены вторым началом.
|
|


|
Часть первая. Каждая термодинамическая система обладает функцией состояния, называемой энтропией. Энтропия вычисляется следующим образом. Система переводится из произвольно выбранного начального состояния в соответствующее конечное состояние через последовательность состояний равновесия. Вычисляются все подводимые при этом к системе порции тепла 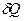 , делятся каждая на соответствующую ей абсолютную температуру Т и все полученные таким образом значения суммируются.
, делятся каждая на соответствующую ей абсолютную температуру Т и все полученные таким образом значения суммируются.
 (13)
(13)
Часть вторая. При реальных (не идеальных) процессах энтропия замкнутой системы возрастает.
Содержание второй части формулировки А.Зоммерфельда в большинстве учебников рассматривается как самодостаточная формулировка второго начала, или как закон возрастания энтропии:
 (14)
(14)
Энтропия не убывает только в процессах изолированной системы, в неизолированной системе энтропия может и возрастать, и убывать, и оставаться неизменной. Рост энтропии в изолированной системе означает приближение системы к состоянию термодинамического равновесия; в этом состоянии S – максимальна, а dS=0. 
Расчет изменения энтропии в различных процессах изучаемых систем зачастую является актуальной внутренней подзадачей разнообразных задач термодинамики, химии биологии, лингвистики. В частности, изменение энтропии служит мерой изменения качества энергии. Важнейшим условием для тепловых машин, работающих по произвольному циклу, является условие их максимально допустимой эффективности:
 (за цикл) (15)
(за цикл) (15)
Большинство процессов, происходящих в природе, необратимы, например: диффузия, расширение, растворение. Для таких процессов вычисление энтропии основывается на том, что S – функция состояния. Если система перешла из одного состояния в другое необратимым образом, то можно мысленно заменить необратимый процесс обратимым, причем начальное и конечное состояния этого процесса должны быть равновесны, рассчитанное в этом случае изменение энтропии будет равно изменению энтропии при реальном необратимом процессе.

Задачи
3.1. Произвольное рабочее вещество совершает цикл, в пределах которого абсолютная температура изменяется в α раз. Цикл имеет вид изображенный на рис.15: T – температура, S – энтропия. Найти КПД цикла.
|
3.3. Найти приращение энтропии алюминиевого бруска массы m=3.0кг при нагревании его от Т1=300˚К до Т2=600˚К, если в этом интервале температур удельная теплоемкость алюминия с=а+bТ, где а=0,77Дж/гК, b=0.46мДж/гК2.
3.4. Вычислить изменения внутренней энергии и энтропии одного моля идеального газа при расширении по политропе pVn=const от объема V1 до объема V2. рассмотреть частные случаи изотермического и адиабатического процессов.
3.5. Гелий массы m=1,7г адиабатически расширили в n=3 раза и затем изобарически сжали до первоначального объема. Найти приращение энтропии газа в этом процессе.
3.6. В двух сосудах одного и того же объема находятся различные идеальные газы. Масса газа в первом сосуде М1, во втором М2, давления газов и их температуры одинаковы. Сосуды соединили друг с другом и начался процесс диффузии. Определить суммарное изменение ∆S энтропии рассматриваемой системы, если относительная молекулярная масса первого газа μ1, а второго μ2.
 |
3.7. Идеальный одноатомный газ в количестве υ=10 молей, находящийся при температуре Т1=300˚К, расширяется без подвода и отдачи тепла в пустой сосуд через турбину, необратимым образом совершая работу (рис.16) После установления равновесия температура газа понижается до Т=200˚К.
|
|
3.8. Вычислить изменение энтропии при смешении одноатомного идеального газа массы m1, имеющего начальную температуру Т1 и давление p1, и двухатомного газа массы m2, имеющего начальные температуру Т2 и давление p2. Молярные массы смешиваемых газов М1 и М2.
Ответы
3.1. 
3.2. 
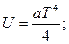
3.3. 
3.4. 

где k=const;
3.5. 
3.6. 
3.7. 
3.8. 
Семинар 4. Реальные газы
Уравнение состояния газа Клапейрона-Менделеева имеет ограниченную область применимости, поскольку не учитывает межмолекулярные взаимодействия. В реальных газах есть дальнодействующие силы притяжения и короткодействующие силы отталкивания. Взаимодействие в газах приводит к количественным и качественным отклонениям от поведения, предсказываемого уравнением Клапейрона-Менделеева.
 Существует множество уравнений реальных газов, адекватно описывающих их свойства, включая превращение в жидкость. Это уравнения Ван-дер-Ваальса, Дитеричи (16), Бертло (17), Клаузиуса (18). Наиболее популярные в современной научной практике уравнения Редлиха-Квонга, Пенга-Робинсона, Камерлинг-Оннеса, или вириальное уравнение (19).
Существует множество уравнений реальных газов, адекватно описывающих их свойства, включая превращение в жидкость. Это уравнения Ван-дер-Ваальса, Дитеричи (16), Бертло (17), Клаузиуса (18). Наиболее популярные в современной научной практике уравнения Редлиха-Квонга, Пенга-Робинсона, Камерлинг-Оннеса, или вириальное уравнение (19).
|
 , функцией расстояния r между центрами молекул (рис.17).
, функцией расстояния r между центрами молекул (рис.17).
| Уравнения состояния реальных газов |
 - уравнение Дитеричи (16), - уравнение Дитеричи (16),
 - уравнение Бертло (17), - уравнение Бертло (17),
 - уравнение Клаузиуса (18), - уравнение Клаузиуса (18),
 - уравнение Камерлинг-Онесса (19). - уравнение Камерлинг-Онесса (19).
|
Функция имеет минимум, в котором силы притяжения уравновешиваются силами отталкивания. Аналитический вид функции на полуэмпирической основе представлен ниже:
 - потенциал Леннарда-Джонса (20)
- потенциал Леннарда-Джонса (20)
В теории Ван-дер-Ваальса используется упрощенная модель межмолекулярного взаимодействия, часть кривой заменяется вертикальной прямой (пунктирная линия на рис.17). Если d – расстояние до этой прямой от начала координат, то центры взаимодействия частиц не могут сблизиться на расстояние, меньшее d, что соответствует модели твердых упругих шаров.
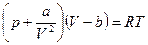 - уравнение Ван-дер-Ваальса (21)
- уравнение Ван-дер-Ваальса (21)
где 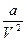 – поправка на давление, т.е. дополнительное «внутреннее» давление за счет взаимного притяжения; b – поправка на объем молекул, учитывающая силы отталкивания.
– поправка на давление, т.е. дополнительное «внутреннее» давление за счет взаимного притяжения; b – поправка на объем молекул, учитывающая силы отталкивания.
 Наиболее содержательные результаты получаются из уравнения Ван-дер-Ваальса путем анализа его изотерм. Если в уравнении (19) принять Т = const, то изотерма этого уравнения в плоскости p, V пересекается прямой линией p = const либо в одной точке, либо в трех точках (рис.18). При некоторой промежуточной температуре Ткр три корня V1, V2, V3 становятся равными.
Наиболее содержательные результаты получаются из уравнения Ван-дер-Ваальса путем анализа его изотерм. Если в уравнении (19) принять Т = const, то изотерма этого уравнения в плоскости p, V пересекается прямой линией p = const либо в одной точке, либо в трех точках (рис.18). При некоторой промежуточной температуре Ткр три корня V1, V2, V3 становятся равными.
Такая температура и соответствующая ей изотерма называются критическими. Критическая изотерма всюду монотонно опускается вниз, за исключением одной точки – критической.
Соответствующие этой точке давление pk, объем Vk и температура Tk также называются критическими.
|

