




Рассмотренные выше классические представления о растворах электролитов можно охарактеризовать как континуальные модели, в которых ионы представляются в виде заряженных жестких сфер, погруженных в непрерывную диэлектрическую среду (континуум). Такие модели называют иногда «примитивными». Однако при внесении в них целого ряда физически обоснованных уточнений удается получить результаты, близкие к реальности.
Наиболее принципиальным недостатком «примитивных» моделей является игнорирование пространственной дисперсии диэлектрической проницаемости, т.е. локального снижения диэлектрической проницаемости в ближайшей окрестности ионов, в первом приближении — в пределах первой сольватной оболочки. Ответственными за пространственную дисперсию в протонных растворителях являются, как уже отмечалось выше, диэлектрическое насыщение при упорядочении ближайших диполей в сильном электрическом поле иона и разрушение структуры растворителя в результате вовлечения части молекул в сольватные оболочки. Для ряда растворителей в случае преобладания эффектов диэлектрического насыщения пространственное распределение диэлектрической проницаемости можно удовлетворительно описать ступенчатой зависимостью: она равна ε = εоп в пределах первой сольватной оболочки и скачком увеличивается до ε=ε′ при больших расстояниях от центра иона (рис. 3.3). Для протонных растворителей, в том числе воды, вследствие особенностей процессов релаксации картина, по-видимому, существенно сложнее, а в пределах первой сольватной оболочки величина диэлектрической проницаемости близка к ε при частотах инфракрасной области.
При усовершенствовании континуальных моделей неизбежно возникают эмпирические модельные параметры, но без таких параметров не удается обойтись и в более совершенных подходах. Основной задачей является, вообще говоря, не расчет параметров, а описание общего вида зависимостей наблюдаемых термодинамических свойств от температуры, концентрации, давления, состава и т. д.
Так, с учетом пространственной дисперсии диэлектрической проницаемости для энергии сольватации вместо уравнения (2.3.5) можно получить:
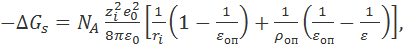 (3.6.1)
(3.6.1)
где ρоп - радиус корреляции для ориентационной поляризации, близкий в первом приближении к толщине сольватной оболочки.
Это уравнение дает более близкие к эксперименту значения ∆Gs. С учетом пространственной дисперсии ε можно обосновать и частотную дисперсию диэлектрической проницаемости, и зависимости этой величины от концентрации электролита.
«Непримитивные» модели, в которых растворитель и ионы электролита рассматриваются в одинаковом микроскопическом масштабе, можно условно разделить на две группы. Первая характеризуется рассмотрением жидких фаз как сильно разупорядоченных кристаллов (дальний порядок в которых существует, как правило, на расстояниях около 4–5 молекулярных диаметров). Вторая группа моделей основывается на описании жидкостей как сильно неидеальных газов. В «непримитивных» моделях молекулы растворителя аппроксимируют твердыми сферами, являющимися диполями, или точечными диполями.
Развитие моделей неидеальных газов в рамках статистической механики основано на рассмотрении бинарных (парных) корреляционных функций, которые можно прямо сопоставлять с экспериментальными данными дифракционных методов.
Парная корреляционная функция g(r) — это нормированная на плотность вещества вероятность найти частицу в точке с координатой r при фиксировании (в точке с нулевой координатой) другой, взаимодействующей с ней частицы. Такая функция является характеристикой локальной плотности и связана некоторым интегральным уравнением с потенциальной энергией системы U. В свою очередь, U является суммой энергий всех парных, тройных и более сложных взаимодействий, причем вклад парных взаимодействий является преобладающим. Решение проблемы взаимосвязи энергий парного взаимодействия u(r) и потенциальной энергии системы U и является основным предметом статистико-механической теории растворов.
При построении парных корреляционных функций необходим учет так называемых «непрямых взаимодействий». Таковыми являются взаимодействия одной из частиц пары с любой третьей частицей, формирующиеся в результате влияния на последнюю второй из частиц рассматриваемой пары. Учет таких взаимодействий, выраженных через параметры зависимости u(r), приводит к необходимости решать сложные системы интегральных уравнений. В отсутствие каких-либо упрощений [подход, в зарубежной литературе называемый «hypernetted chain approximation» (HNС)] интегральные уравнения могут быть решены только численно.
Имеются ограничения по размерам моделируемого ансамбля, так как даже современные быстродействующие компьютеры позволяют решать уравнения HNC лишь для систем, содержащих не более тысячи атомов. Много полезных результатов было получено, однако, и при анализе ансамблей меньшего размера.
Статистико-термодинамическое рассмотрение предполагает, что рассматриваемый ансамбль частиц является каноническим, т.е. среднее значение любой функции f(r) — <f> — от всех координат r составляющих его частиц может быть найдено по формуле
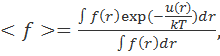 (3.6.2)
(3.6.2)
где интегрирование проводится по всему объему системы (конфигурационному пространству). Иными словами, все невзаимодействующие системы в бесконечном ансамбле находятся в одном и том же термодинамичеcком состоянии и различаются только на микроскопическом уровне. С учетом фундаментального постулата статистической термодинамики об эквивалентности усреднения по времени и по ансамблю можно говорить о том, что все системы в ансамбле имеют равные объемы, температуры и включают одинаковое число частиц.
Существуют различные подходы к переходу от рассчитываемых в «непримитивных» моделях потенциальных энергий к измеряемым термодинамическим величинам. Уравнение состояния при описании системы в терминах парных взаимодействий имеет вид:
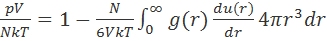 . (3.6.3)
. (3.6.3)
На основе уравнения (3.6.3) можно рассчитать давление, а затем, исходя из термодинамического соотношения
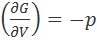 , (3.6.4)
, (3.6.4)
определить свободную энергию Гиббса интегрированием давления по объему при постоянной температуре.
Для решения интегральных уравнений HNC применяется метод Монте-Карло (МК), основанный на нахождении минимума потенциальной энергии канонического ансамбля при заданных постоянных объеме и температуре. Парный потенциал i-й и j-й частиц в приближении твердых сфер равен бесконечности внутри иона (парная корреляционная функция равна нулю), а при расстояниях, больших радиусов ионов, обычно выражается как кулоновский с поправкой на ван-дер-ваальсовы взаимодействия.
Расчеты методом МК в целом хорошо описывают периодический характер распределения ионов в растворе (размытая кристаллическая решетка). Зависимость средних коэффициентов активности от концентрации в области средних концентраций получается близкой к «закону корня кубического».
Метод молекулярной динамики (МД), получивший развитие при достижении достаточно высокого быстродействия компьютеров, предполагает задание наряду с парными потенциалами также скоростей всех частиц ансамбля с постоянными N, Vи полной энергией (микроканонический ансамбль). На основе численного решения системы дифференциальных уравнений ньютоновской механики проводят расчет мгновенных скоростей и траекторий движения взаимодействующих частиц. В результате температуру системы можно вычислить усреднением кинетической энергии по времени. Наряду со структурными характеристиками метод МД позволяет получать также динамические характеристики ансамбля и рассчитывать из них вязкости, коэффициенты диффузии и другие величины.
Очевидно, что оба рассмотренных метода компьютерного моделирования — МК и МД (которые, разумеется, применимы и вне рамок HNC, т.е. для более простых моделей) — наряду с проблемами стоимости и продолжительности расчета имеют существенные ограничения и методические недостатки. Требует специального решения проблема фиксации границ ячейки, в которую заключены частицы ансамбля. Ограничение на количество частиц приводит к недостаточности рассматриваемых ансамблей при моделировании растворов реального состава. Наиболее результативны поэтому расчеты для чистых растворителей, а также для кластеров, образуемых единичным ионом и большим (100 и более) числом молекул воды (модель разбавленных растворов).
В методе МД движение частиц молекулярного и атомного размеров описывается законами классической механики, что некорректно для частиц малых масс и размеров. Перспективы развития метода связаны с учетом квантовых эффектов движения частиц.
Еще одним современным направлением является сочетание методов МД и МК с квантово-химическими расчетами высокого уровня. Наиболее продвинуты ab initio расчеты комплексов металлов с водой и другими растворителями. Эти системы могут рассматриваться как модели ионов с ближайшей сольватной оболочкой.
Сочетание различных методов позволяет устранить еще одно ограничение метода МД, возникающее при обычно принимаемом допущении об аддитивности парных потенциалов. В системах, в которых природа частиц изменяется (например, в результате ассоциации или химических превращений), ab initio расчеты обеспечивают возможность подстраивать изменяющиеся во времени парные потенциалы в ходе компьютерного моделирования (Р. Кар и М. Парринелло, 1985 г.). По-видимому, только компьютерное моделирование позволит в будущем решить проблему описания растворов полиатомных частиц сложного строения.
Отмеченные проблемы компьютерного моделирования не позволяют пока отказаться от более простых подходов к описанию растворов электролитов. Так, широко используется упрощенный вариант HNC — так называемое среднесферическое приближение (mean spherical approximation, MSA), позволяющее дать аналитическое решение систем интегральных уравнений, построенных в рамках подхода на основе коррелятивных функций Боголюбова.
В первоначальном варианте теории МSA ионы и диполи растворителя рассматриваются как твердые сферы определенного размера. Моменты высших порядков (например, квадрупольные) игнорируются, а химический состав системы никак не учитывается. Три типа коррелятивных функций, описывающих ион-ионные, ион-дипольные и диполь-дипольные взаимодействия, строятся в предположении о чисто электростатической природе всех взаимодействий. При описании свойств протонных растворителей, не содержащих ионов, MSA позволяет ограничиться введением одного эмпирического параметра — так называемого «параметра прилипания», описывающего дополнительное направленное взаимодействие (водородную связь). Это возможно, однако, только при условии слабо выраженной несферичности молекул растворителя. Для воды данное условие неплохо выполняется, параметр rs для воды принимают обычно равным 0,142 нм.
MSA дает поправку к уравнению Дебая-Хюккеля для среднего коэффициента активности (3.3.1) в виде дополнительного слагаемого в правой части, зависящего только от собственных размеров ионов (контактного расстояния а). Поправка обеспечивает согласие с экспериментальными данными для 1,1-электролитов при концентрациях до 0,3 М и дает завышенные значения
для более концентрированных растворов. При учете эффективной зависимости диэлектрической проницаемости от концентрации уравнение MSA работает вплоть до концентраций 1 М.
Существенно, что аналогичное введение ε(c) в уравнение Дебая-Хюккеля практически не влияет на верхний концентрационный предел его применимости.
Метод MSA может быть использован также для уточнения уравнения Фуосса [уравнение (3.4.2)] и нахождения поправки к нему для растворов с высокой ионной силой. Наибольшее развитие в электрохимии он получил применительно к электростатическим задачам в гомогенной и электрохимической кинетике.
Перспективы развития теории растворов в значительной степени связаны с темпами роста возможностей и быстродействия компьютерной техники. Не менее важны и дополнительные экспериментальные данные о микроскопическом строении растворов, которые позволили бы уточнять параметры теории. Вообще говоря, необходимо руководствоваться как минимум следующими критериями достоверности теории: согласие с опытными зависимостями свойств раствора электролита от концентрации и температуры; возможность описания свойств многокомпонентных растворов; возможность согласованного описания термодинамических и неравновесных свойств растворов.
Список литературы
1. Электрохимия / Б. Б. Дамаскин, О. А. Петрий, Г. А. Цирлина. — 2 изд., испр. и перераб. — М.: Химия, «КолосС», 2006. —672 с.: ил. — (Учебники и учеб. пособия для студентов высш.учеб. заведений)
2. РАСТВОРЫ ЭЛЕКТРОЛИТОВ / Р.Робинсон,Р.Стокс -М.: Издательство иностранной литературы, 1963. -646 с.
3. Успехи физических наук / Г.А. Мартынов/ - 1967., том 91, вып. 3 с 455-483.

