




К средним циклам относятся алициклические соединения, содержащие 7-12 звеньев. Макроциклы содержат 12и более звеньев.
Большинство методов получения малых и обычных циклов для получения циклов среднего и большого размеров малоэффективны. Наиболее распространенным способом является их синтез по Ружичке прокаливанием ториевых солей двухосновных кислот в вакууме
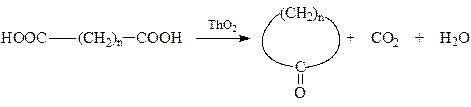
Другой путь, используемый для получения среднециклических кетонов - это реакции сложноэфирной конденсации по Дикману или конденсации нитрилов по Торпу. Однако эти способы неприменимы для получения 9- и 10-членных циклов. Основной проблемой синтеза средних и больших циклов является протекание межмолекулярной реакции конденсации вследствие падения вероятности столкновения концов одной молекулы. Одним из путей решения этой проблемы является сильное разбавление реакционной смеси, направленное на уменьшение возможности протекания межмолекулярных реакций.
Наиболее эффективным путем для получения циклических соединений среднего и большого размеров является ацилоиновая конденсация
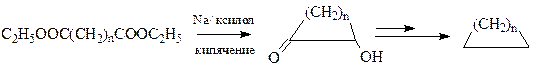
Образующийся ацилоны затем можно перевести в необходимый продукт, в том числе алицикл в процессе восстановительного элиминирования.
Современным методом получения макроциклических соединений с использованием так называемого «низковалентного» титана по Макмури является внутримолекулярная циклизация дикарбонильных соединений с получением олефина
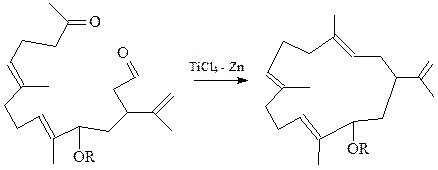
Кроме того, макроциклические соединения получаются на основе реакции метатезиса с использованием катализатора Граббса.
Производные циклогептана
Производные циклогептана получаются методами, используемыми для синтеза обычных циклов.
Из оригинальных способов получения можно отметить реакцию внедрения в молекулу бензола карбена, образующегося в результате распада диазосоединения; пиролиз промежуточного норкарана приводит к производному циклогептатриена
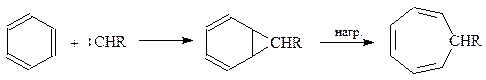
Полученное производное весьма реакционноспособно и в свободном виде легко полимеризуется. Его хлорпроизводное способно генерировать ароматическую систему, образуя соль тропилия
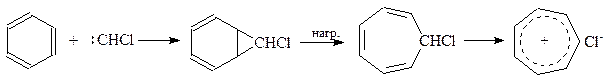
Производное циклооктана
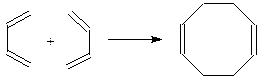
Производные циклооктана также могут быть получены методами синтеза обычных циклов. Его непредельные производные получаются путем димеризации бутадиена на катализаторе Циглера [(Al(C2H5)3 + TiCl3]
Циклооктатетраен получается конденсацией ацетилена по Реппе при нагревании под давлением в присутствии цианида калия
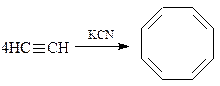
При воздействии ультрафиолетового света эфиры ацетилендикарбоновой кислоты взаимодействуют с бензолом по реакции [2+2]-присоединения и дают производные циклооктадиена
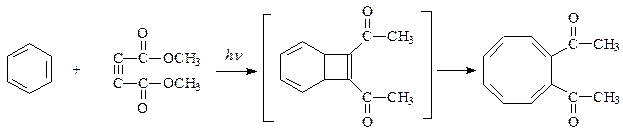
Непредельные производные циклооктана исчерпывающе гидрируются над никелем Ренея, Pt или Pd до циклооктана. При прерывании реакции можно выделять из реакционной смеси все промежуточные продукты гидрирования – циклооктатриен, циклооктадиен, циклооктен и циклооктан.
Циклооктатетраен интересен тем, что содержит в своей структуре 8 углеродных атомов и 4 двойные связи и по своему графическому изображению формально напоминает бензол. Но это не так. В отличие от бензола он имеет неплоскостное строение, а самое главное, согласно правилу Хюккеля не является ароматическим, так как по формуле 4n+2 должен содержать либо 6, либо 10 π -электронов. Следовательно, чтобы придать циклооктатетраену ароматичность необходимо подать в цикл 2 электрона. Действительно, циклооктатетраен реагирует с металлическим калием и приводит к стабильной соли 2К+[C8H8]2-. По своему химическому поведению он похож на алкены: обесцвечивает холодный раствор KMnO4, реагирует с Br2/CCl4. Циклооктатетраен подвергается фотохимическому распаду с образованием бензола и ацетилена. Эти реакции позволяют прийти к выводу о строении реальной молекулы как усредненной форме 3-ёх резонансных структур
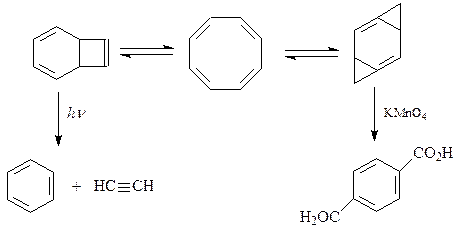
Макроциклы
К ним относятся алициклические соединения, состоящие из 12 углеродных атомов.
Многие природные макроциклические соединения являются биологически активными веществами, поэтому представляют большой практический интерес. Все высшие кетоны обладают характерным запахом. Кетоны с 10, 11 и 12 атомами в цикле пахнут камфорой, с 14-18 атомами – мускусом. За способность удерживать молекулы душистого вещества экзальтон, мускон, цибетон и другие высшие кетоны используются в парфюмерии в качестве стабилизаторов запахов
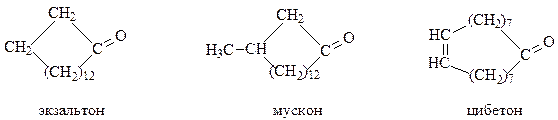
Каркасные алициклы
Помимо мостиковых циклических соединений известны объемные циклические системы, в которых каждый из циклов сконденсирован с тремя или четырьмя соседними, например
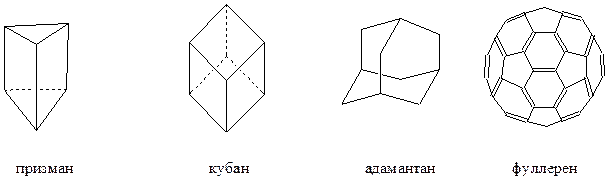
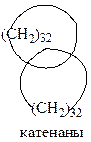
Фуллерены – соединения, состоящие исключительно из атомов углерода, соединенных в конденсированную поликарбоциклическую систему каркасного типа, состоящую из 12 пятизвенных циклов и нескольких шестизвенных.
Интересный ряд бициклических систем представляют собой катенаны. Катенаны отличаются от других бициклических алканов тем, что кольца в них связаны не валентными связями, а свободно ассоциированы, как звенья в цепи.
Разумеется, для подобных циклических соединений нет общих методов получения и в каждом случае они разрабатываются целенаправленно для конкретного объекта. Например, адамантан может быть получен изомеризацией полностью гидрированного димера циклопентадиена под действием AlCl3
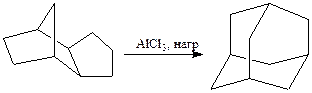
Терпеноиды
Большую группу природных производных алициклических соединений представляют терпеноиды. Эти соединения выполняют важные биологические функции в живых организмах и растениях. Много лет назад было обнаружено, что активное начало запаха можно выделить из растений путем осторожного нагревания и конденсации пара на холодной поверхности. Выделяемые масла стали называть эфирными, из которых в последующем были выделены первые терпены (от немецкого terpentine). Терпены широко распространены в природе, они обнаружены не только в растениях, животных, но и в морских продуктах.
Структурное сходство многих монотерпеноидов и образование изопрена при их термическом разложении позволило предположить, что пятиуглеродный фрагмент является общей структурной единицей всех монотерпеноидов. Это структурное единство позволило Ружичке сформулировать в 1921 году известное «изопреновое правило», согласно которому углеродный скелет терпеноидов состоит из изопреновых фрагментов, связанных в определенном порядке. По мере накопления сведений о терпеноидах стали известны и примеры отступления от «изопренового» правила. Действительно, в результате метаболизма вполне допустимы перегруппировки, которые и могут приводить к отступлению от этого правила. Поэтому считается, что терпеноидами являются соединения, изначально образованные комбинацией изопреновых фрагментов, в результате которой возникают ациклические предшественники: гераниол, фарнезол,геранилгераниол, сквален и другие соединения. Таким образом, терпеноиды делятся на монотерпеноиды, в основе которых лежит С-10 углеродный скелет, сесквитерпеноиды (С-15), дитерпеноиды (С-20) и тритерпеноиды (С-30).
Все монотерпеновые соединения можно разделить на 3 основные группы: ациклические, моно-, би-, трициклические. Из ациклических моно- и сесквитерпеноидов в природе чаще всего встречаются соединения с 2,6-диметилоктановым скелетом
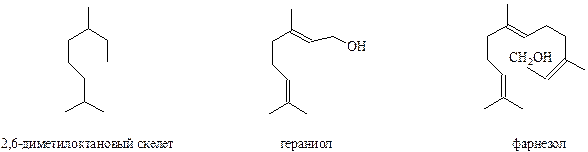
Гераниол и фарнезол широко распространены в природе и обнаружены в цитронелловом, розовом, апельсиновом и многих других эфирных маслах.
Наиболее интересны циклические терпеноиды. К моноциклическим терпеноидам относятся α, β -фелландрен и лимонен
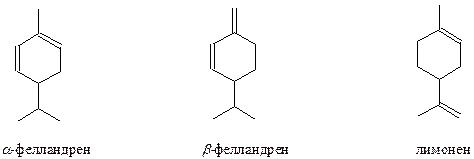
Из бициклических монотерпеноидов наиболее распространены в природе α, β -пинены, ∆ 3-карен:
Аналогичным образом сесквитерпеноиды могут быть моно-, би- и трициклическими. Моноциклические сесквитерпеноиды α - и γ -гумулены содержат 11-членный цикл

В живицах хвойных деревьев найдены соединения группы гермакрана, содержащие 10-членный цикл

Бициклические сесквитерпеноиды широко представлены группой кадинана и другими группами, представляющими структуры, эпимерные кадинанам
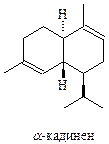
Среди бициклических терпеноидов известны представители, содержащие аннелированные циклопентановые и циклогексановые, циклодекановые и циклобутановые фрагменты.
Группа лонгифолана представляет трициклические терпеноиды. Тетрациклические сесквитерпеноиды содержатся в смолах различных видов хвойных растений.
Большой класс биологически активных дитерпеноидов, обнаруженных на земле, а также выделенных из морских источников представляют многочисленные производные цембрана
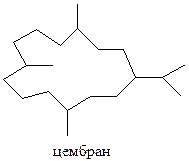
По сравнению с цембраноидами бициклические дитерпеноиды лабданового ряда представлены значительно большим числом соединений
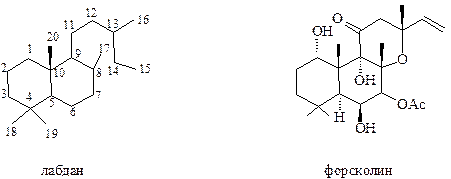
Один из производных лабдана, обладающий понижающим кровяное давление (гипертензивным) действием и эффективным современным препаратом для лечения глаукомы, является форсколин, впервые выделенный из растения, произрастающего в Индии.
К дитерпеноидам относятся новые уникальные по своим биологическим свойствам цитотоксические агенты таксол, элеутеробин, эпотилоны. На их основе созданы или находятся на стадии разработки эффективные противораковые лекарственные препараты
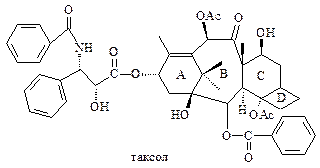
Таксол представляет собой тетрациклический дитерпеноид, обнаруженный в 1962 году в коре тиса Texus brevifolia.
Форсколин является примером того, что бициклические дитерпеноиды в ходе ферментативных трансформаций в биологических объектах, называемых метаболизмом, могут превращаться в трициклические. Аналогичным образом эти превращения могут происходить и между низшими и высшими терпеноидами.
Таким образом, биогенетическую связь между различными классами терпеноидов отражает схема, основанная на последовательной олигомеризации изопентенилпирофосфата
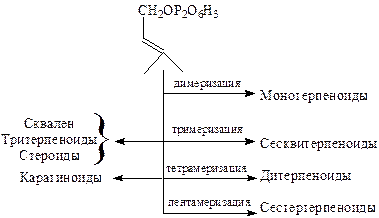
АРОМАТИЧЕСКИЙ РЯД
В органической химии существует огромная группа соединений со сходными и, как когда-то представлялось, необычными и удивительными свойствами. Общим для этих миллионов соединений является не принадлежность к какому-то определенному классу в соответствии с функциональной группой, а наличие в их молекуле стабильной группировки из шести атомов углерода, которая сохраняется при обычных химических реакциях. На это обратил внимание немецкий химик Кекуле еще в начале ХIХ века. Постепенно стало понятно, что между указанной группировкой атомов и с не укладывающимися в привычные рамки свойствами молекул, содержащими эту группировку, существует тесная связь. Сразу же отметим, что упомянутая группировка углеродных атомов получила название бензол, а органические соединения, содержащие остаток бензола в своей молекуле, составили ароматический ряд. Со временем к ароматическому ряду были отнесены и соединения с несколькими бензольными кольцами, а также соединения без бензольных колец, однако близкие к ним по своим свойствам. Отличительные свойства соединений ароматического ряда: значительная устойчивость к действию высоких температур и окислителей и легкость вступления в реакции замещения при формальной ненасыщенности их молекулы, получили обозначение как ароматический характер. Первые известные представители соединений рассматриваемого ряда имели характерный приятный запах (бальзамы, смолы и эссенции). Отсюда и возникло их общее обозначение как ароматический ряд.
Глава 11
Арены
Строение бензола
Бензол впервые был выделен М. Фарадеем в 1825 году из конденсата, выпавшего из светильного газа, используемого для освещения городских улиц Лондона. Фарадей назвал это жидкое легкоподвижное вещество с резким запахом «карбюрированным водородом» (carburated hydrogen). Важно при этом отметить, что уже тогда было установлено, что бензол состоит из равных частей углерода и водорода.
Несколько позже, в 1834 году, Митчерлих получил бензол декарбоксилированием бензойной кислоты. Он же установил элементный состав полученного соединения – С6Н6 – и предложил свое название для него – бензин. Однако с этим названием не согласился Либих. Ему показалось, что это название ставит бензол в один ряд с такими далекими от него веществами как хинин и стрихнин. По мнению Либиха более удачным названием для нового соединения является бензол, посколькуоно показывает близость бензола по свойствам к маслам (от немецкого оl – масло). Были и другие предложения. Поскольку бензол был выделен Фарадеем из светильного газа, то Лоран предложил (1837 г.) для него название фено от греческого «несущий свет». Это название не утвердилось, однако именно от него произошло название одновалентного остатка бензола – фенил.
Углеводороду Фарадея не повезло. Все предложенные для него названия оказались ущербными. Из либиховского названия «бензол» следует, что соединение содержит гидроксильную группу, которой там нет. Точно так же митчерлиховский «бензин» не содержит функциональную азотсодержащую группу. Более того, существование различных названий привело к разделению химиков. В немецкой и русской научной литературе утвердилось название «бензол», а в английской и французской – «бензен» (bensene, toluene, xylene).
На первый взгляд кажется, что установить строение бензола не представляет больших трудностей. В состав молекулы бензола входит лишь два элемента, на шесть атомов углерода приходится шесть атомов водорода. К тому же физические и химические свойства бензола изучены весьма обстоятельно. Однако эта работа затянулась на многие десятилетия и завершилась лишь в 1931 году.
Наиболее трудные барьеры к познанию структуры бензола были преодолены выдающимся немецким химиком Кекуле. С высоты современных знаний трудно понять и оценить значение выдвинутой им гипотезы, согласно которой молекула бензола имеет циклическое строение (1865 г.). Однако именно это предположение, при совокупном рассмотрении с числом изомеров у моно- и дизамещенных бензолов, привело Кекуле к известной формуле. По Кекуле бензол представляет собой шестичленное циклическое соединение с тремя чередующимися двойными связями, т.е. циклогексатриен
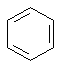
Именно эта структура согласуется с существованием одного и только одного монозамещенного бензола и трех изомеров дизамещенных бензолов
C момента появления структуры Кекуле началась ее критика, которую она, к сожалению, вполне заслуживала. Уже отмечалось, что характерная особенность ароматических соединений - присущий им ароматический характер. Структура Кекуле для бензола оказалась не в состоянии объяснить эту особенность ароматических соединений. В ряде случаев она не могла также объяснить отсутствия изомеров, в то время как формула циклогексатриена для бензола допускала их существование. Так, орто -замещенных бензолов может быть два изомера
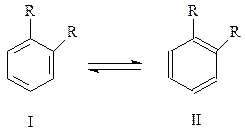
однако обнаружить их не удалось. Сразу же отметим, что для преодоления этого затруднения Кекуле предложил рассматривать бензол как циклогексатриен с подвижными, не закрепленными, двойными связями. В результате быстрого преобразования I во II и наоборот бензол ведет себя как структура как бы состоящая из равных количеств I и II.
Итак, основной недостаток бензола Кекуле – невозможность объяснить на его основе ароматический характер соединений, содержащих в своей молекуле бензольное кольцо. Если бы бензол был циклогексатриеном, т.е. соединением с тремя двойными связями, то он должен был бы:
- легко окисляться даже холодным водным раствором КMnO4,
- уже при комнатной температуре присоединять бром и легко вступать в другие реакции электрофильного присоединения,
- быстро гидрироваться водородом в присутствии никеля при комнатной температуре,
В эти реакции бензол вступает неохотно, не так как алкены. А вот реакции замещения - весьма характерны для соединений ароматического ряда. Отсюда следует, что бензол не может быть циклогексатриеном и формула Кекуле не отражает истинного строения бензола. Основной недостаток бензола Кекуле – присутствие в нем двойных связей. Если бы их не было, то и не следовало бы ожидать от бензола проявления свойств, характерных для алкенов. В этой связи становится понятным, почему все дальнейшие попытки «усовершенствовать» формулу Кекуле носили форму лишить ее двойных связей, сохранив при этом за бензолом циклическое строение. Таковы формулы III – VII, предложенные Клаусом (1867 г.), Дьюаром (1867 г.), Армстронгом – Байером (1887 г.), Тиле (1899 г.) и Ладенбургом (1869 г.)
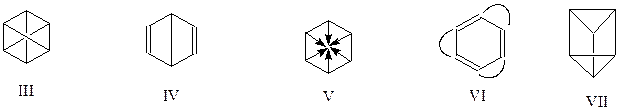
Ни одна из этих формул не могла объяснить всех свойств, присущих бензолу. Это стало возможным только с развитием квантовой химии.
Согласно современным представлениям о строении бензола его молекула представляет собой плоский правильный шестиугольник, на вершинах которого расположены углеродные атомы, находящиеся в sp 2–гибридном состоянии. Каждый из шести углеродных атомов за счет трех тригональных гибридных орбиталей образует две σ -связи с соседними углеродами и еще одну связь с водородом. Все эти связи расположены в одной плоскости под углом 1200 друг к другу. В гибридизации участвуют лишь два из трех р -электронов углеродных атомов. Поэтому после образования σ -связей у каждого из шести углеродов бензольного кольца остается еще по одному р -электрону. Из истории установления строения бензола, растянувшегося на многие десятилетия, видно насколько трудно пробивало себе дорогу представление, что р -электроны способны перекрываться друг с другом не только попарно с образованием π -связей. При некоторых обстоятельствах возможно перекрывание облаков р- электронов как с соседом справа, так и с соседом слева
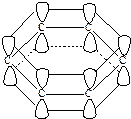
Это становится возможным, если молекула имеет циклическое строение, расстояния между углеродами одинаковы и оси р -электронов параллельны между собой. Последнее условие соблюдается, если молекула имеет плоское строение.
При таком построении молекулы бензола углеродные атомы связаны между собой не ординарными и не двойными связями. Эти связи, скорее всего, следовало бы отнести к «полуторным». Не лишним будет упомянуть, что согласно результатам рентгеноструктурного анализа кристаллического бензола все углерод-углеродные связи в бензоле имеют одинаковую длину 0.14 нм, которая является промежуточной между простой (0,154 нм) и двойной (0,134 нм) связями.
Таким образом, согласно современным представлениям в бензоле нет типичных двойных связей между углеродами. Следовательно, от такого соединения и не следует ожидать проявления свойств, обусловленных двойными связями. В то же время нельзя отрицать значительной непредельности молекулы бензола. Циклоалкан с шестью углеродами (циклогексан) содержит 12 водородных атомов, в то время как у бензола их всего 6. Отсюда следует, что формально бензол мог бы иметь три двойные связи и в реакциях присоединения вести себя как циклотриен. Действительно, в условиях реакций присоединения бензол присоединяет по три молекулы водорода, галогенов или озона.
В настоящее время в научно-технической литературе используется два графических изображения бензола

Одно из них подчеркивает непредельный характер бензола, а другое – его ароматичность.
Как же увязать строение бензола с его характерными свойствами, главным образом, с его ароматическим характером? Почему бензол проявляет уникальную термодинамическую устойчивость?
В свое время было показано, что алкены довольно легко присоединяют молекулу водорода и превращаются в алканы. Эта реакция протекает с выделением тепла, около 125,61 кДж на каждую двойную связь, и носит название - теплоты гидрирования. Попробуем использовать теплоту гидрирования для оценки термодинамической устойчивости бензола.
Реально существующие циклогексен, циклогексадиен и бензол гидрируются в циклогексан
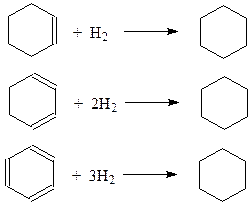
Теплота гидрирования циклогексена составила 119,75 кДж. Тогда ожидаемое значение для циклогексадиена должно составить 119,75 х 2=239,50 кДж (в действительности 231,96 кДж). Если бы бензол имел три двойные связи (циклогексатриен Кекуле), то теплота гидрирования для него должна была бы быть 119,75 х 3=359,25 кДж. Экспериментальное же значение в последнем случае разительно отличается от ожидаемого. При гидрировании бензола выделяется лишь 208.51 кДж тепла, что меньше ожидаемого значения на 359,25 – 208.51=150,73 кДж. Эта энергия носит название энергии резонанса. Если при гидрировании бензола выделяется на 150,73 кДж меньше энергии от ожидаемого значения, то это означает лишь то, что сам бензол уже изначально содержит на 150,73 кДж меньше энергии, чем гипотетический циклогексатриен. Отсюда следует, что бензол не может иметь строения циклогексатриена. Стабильность молекулы бензола на величину энергии резонанса есть результат отсутствия в ней изолированных двойных связей и наличия единого электронного облака секстета р -электронов.
Приобретая благодаря выгодам своего строения высокую термодинамическую устойчивость, бензол в ходе химических реакций всячески стремится сохранить эту устойчивость. Понятно, что это может быть реализовано только при условии сохранения при химической реакции бензольного кольца в неизменном виде. Такую возможность предоставляют лишь реакции замещения и именно по этой причине для соединений ароматического ряда более характерны реакции замещения, чем присоединения. В ходе реакций электрофильного присоединения ароматическое соединение перестает быть ароматическим, теряет исключительную стабильность вместе с энергией резонанса, обуславливающей как раз эту стабильность. По этой причине ароматические соединения вступают в реакции присоединения гораздо труднее, чем, например, алкены. Другая особенность реакций присоединения с участием ароматических соединений – это их бескомпромиссность. Они либо не вступают в реакции присоединения, либо присоединяют сразу все. Об этом свидетельствует тот факт, что не удается получить из бензола продукты частичного гидрирования или хлорирования. Если уже эти реакции идут, то протекают таким образом, что сразу получаются продукты полного гидрирования или хлорирования
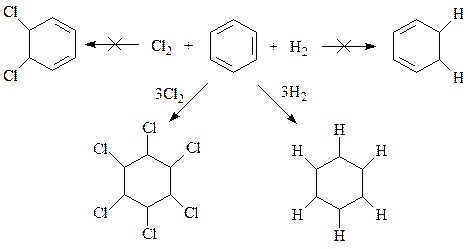
Такое развитие событий связано с тем, что единое электронное облако шести р -электронов у бензола или существует или не существует, промежуточные варианты для него исключены.
Ароматичность
В связи с существованием у некоторых соединений ароматического характера, обусловленного своеобразием их строения, встает вопрос, какие же особенности строения молекулы ответственны за проявления этих свойств?
Из материала предыдущего раздела понятно, что для этого необходимо, чтобы молекула содержала циклическую систему делокализованных р -электронов. По очевидным соображениям, возникновение единого облака таких электронов возможно только в том случае, если молекула циклическая (только в этом случае возможно появление кольцевой системы) и плоская (иначе нарушится параллельность осей р -электронов). Однако оказывается, что все изложенное выше необходимое, но не достаточное условие для проявления соединением ароматического характера. Определяющим фактором оказалось число р -электронов, образующих циклическую систему. У соединений, обладающих ароматическим характером, их в молекуле должно быть строго определенное число: 2, 6, 10, 14 и т.д. Как установил Э.Хюккель, единое электронное облако у ароматической системы должно насчитывать
(4n + 2)
р -электронов, где n=0, 1, 2, 3, 4 и т.д. Эта формула выведена Хюккелем из соображения заполнения различных орбиталей, образующих р -облако, и более подробно в данной книге не рассматривается.
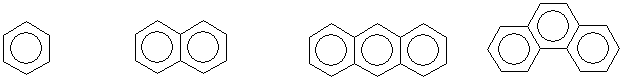
Итак, если молекула циклическая, плоская и содержит хюккелевское число р -электронов, то она будет обладать ароматическим характером, т.е. проявлять высокую термодинамическую устойчивость и легко вступать в реакции замещения при формальной непредельности. Всем указанным требованиям удовлетворяют бензол (n=1), нафталин (n=2), антрацен и фенантрен (n=3) и другие полициклические ароматические системы
Пока вне поля зрения оставался случай с n=0, т.е. система, состоящая из двух р -электронов. Понятно, что два электрона могут составить «единую циклическую систему делокализованных электронов» не при любом размере кольца. Так, два р -электрона в циклобутановом, циклопентановом или же в циклогексановом кольце составят лишь молекулы циклобутена, циклопентена и циклогексена, не проявляющих ароматического характера
Может быть, ароматическим характером будет обладать молекула циклопропена? Оказывается, тоже нет. В молекуле циклопропена невозможна делокализация электронов. Другое дело случай циклопропенил-катиона, когда такая возможность возникает. Действительно, один из производных иона циклопропения С3Н3+, синтезированный по следующей схеме
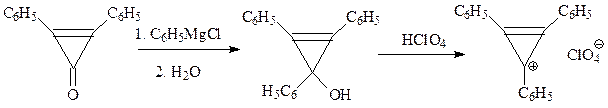
представлял собою достаточно устойчивую при хранении соль (перхлорат), в молекуле которой все углерод-углеродные связи оказались равной длины, а р -электроны сильно делокализованы
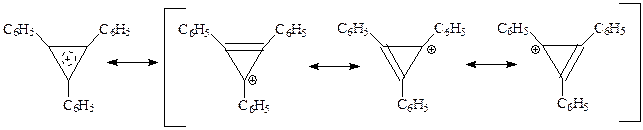
Понятно, что рассматриваемая система обладала ароматическим характером. Следует отметить, что ион циклопропения представляет собой самый маленький цикл, которому присущ ароматический характер. Другая особенность этой системы заключается в том, что ароматический характер проявляет не нейтральная молекула, а ион. В последующем оказалось, что имеются и другие примеры подобного рода.
Циклопентадиен (1) не может быть отнесен к ароматическим системам на том основании, что содержит лишь 4 р -электрона. Это не согласуется с правилом Хюккеля. Однако положение меняется, если молекулу циклопентадиена покинет один из водородов из метиленовой группы, оставив свой электрон. Например, в результате следующей реакции
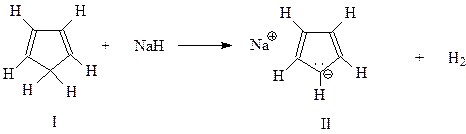
В этом случае возникнет циклопентадиенил-анион II, в котором уже возможно образование замкнутого кольца шести делокализованных р -электронов. В результате циклопентадиенил-анион, в отличие от самого циклопентадиена, должен обладать ароматичностью. Установлено, что этот анион действительно существует в виде дициклопентадиенила железа (ферроцена) III. Ферроцен имеет строение сэндвича, в котором между двумя циклопентадиенил-анионами располагается атом двухвалентного железа
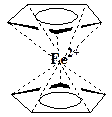
Ферроцен действительно обладает ароматичностью. Все связи между углеродными атомами в циклопентадиенильных анионах выравнены и составляют 0,14 нм. Благодаря приобретению ароматичности, рассматриваемый анион проявляет повышенную устойчивость, о чем свидетельствует его высокая для углеводорода кислотность (Ка =10-15; для сравнения - для циклогептатриена Ка =10-45). Ферроцен вступает также в типичные для ароматических соединений реакции электрофильного замещения: сульфирования, алкилирования и др.
На примере циклопропенил-катиона было уже показано, что ароматичностью может обладать и катион. Другим широко известным примером такого типа ионов может послужить циклогептатриенильный катион, называемый также тропилий-ионом
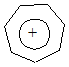
Оказалось, что тропилий-катион представляет собой правильный семиугольник, в котором шесть углеродов имеют по р -электрону, необходимых для образования ароматического секстета. Еще у одного углерода имеется не занятая р -орбиталь, которая обеспечивает возможность делокализации электронов и образования единого электронного облака. Как и следовало ожидать, тропилий-катион, например, в составе бромистого тропилия, обладает ароматическим характером. Он достаточно устойчив и легко вступает в реакции замещения.
Кроме рассмотренных случаев, ароматическая система делокализованных р -электронов может возникнуть и другими способами. Примерами могут послужить хорошо известные пятичленные гетероциклические соединения: фуран, пиррол и тиофен. Казалось бы, что этим соединениям никак не попасть в число ароматических. Хотя их молекулы циклические и имеют плоское строение, однако, у них не хюккелевское число р -электронов – всего 4. В то же время у молекул этих гетероциклов стремление к образованию термодинамически устойчивой ароматической системы настолько велико, что они «добирают» необходимый секстет р -электронов за счет вовлечения в сопряжение неподеленных электронных пар атомов кислорода, азота и серы
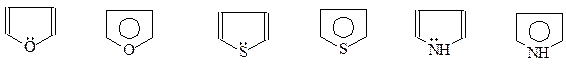
Участие в ароматическом сопряжении неподеленных р -электронов гетероатома легко может быть доказано. Так, пиррол, в отличие от вторичных аминов, не проявляет основности. А когда неподеленные электроны азота перестают участвовать в образовании замкнутой системы р -электронов, как это имеет место в гидрированной молекуле пиррола – пирролидине, то молекула сразу же приобретает основность, присущую вторичным аминам
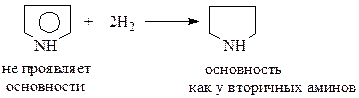
Как и следовало ожидать, пиррол, фуран и тиофен проявляют ароматичность. Подробнее это можно будет увидеть при рассмотрении химии этих соединений.
Наконец, отметим, что способы образования замкнутой системы делокализованных р -электронов отнюдь не исчерпываются рассмотренными примерами. Чтобы показать это рассмотрим молекулы коронена и порфина. Порфиновая система входит в молекулу важнейших природных систем, на которых построена жизнедеятельность живых организмов (гемоглобин) и растений (хлорофилл)
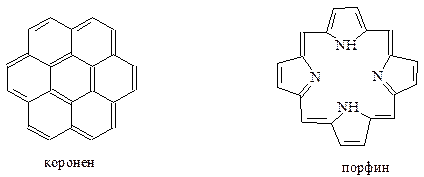
Из вышеприведенной формулы для коронена не следует, что это соединение будет обладать ароматичностью, т.к. не соблюдается требование правила Хюккеля относительно числа р -электронов. Здесь их всего 24, а должно быть 26 (при n=6). Между тем хорошо известно, что коронен проявляет ароматический характер. Каким же образом коронен «сделать» ароматическим по строению его молекулы? Оказывается, проблема решается, если представить коронен как бы состоящим из двух концентрических колец, причем одно из них находится внутри другого. Раздельный подсчет числа р -электронов для каждого из колец позволяет оба кольца отнести к ароматическим. Внутреннее кольцо имеет 6 электронов (n=1), внешнее кольцо включает 18 электронов (n=4). Таким образом, оба кольца содержат хюккелевское число р -электронов и должны быть отнесены к ароматическим системам.
Порфиновая ароматическая система сложена из 4 пиррольных колец, связанных между собой метиновыми группами. Формально в молекуле порфина можно насчитать 11 двойных связей, включающих 22 р -электрона. Кроме них в ароматическом сопряжении участвуют еще 8 р -электронов в составе неподеленных электронных пар у атомов азота. Суммарное число р -электронов 30 удовлетворяет требованиям правила Хюккеля при n=7.
Изомерия и номенклатура
Соединения, молекула которых включает замещенное на алкил - или алкенил бензольное кольцо, называются аренами. Арены, кроме случая бензола, относятся к жирноароматическим.
Ниже приведены наиболее часто встречающиеся арены.
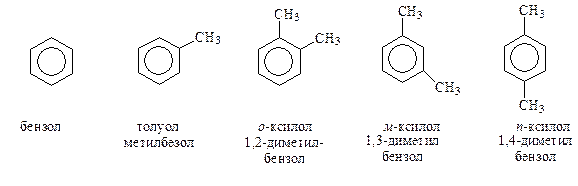
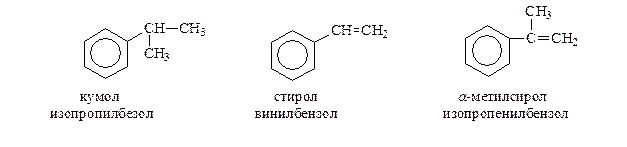
Свое название простейшие арены получают по тривиальной номенклатуре или же как производные бензола. В случае аренов сложного строения название удобнее производить, взяв за основу боковую цепь.
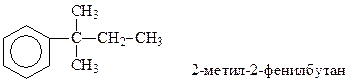
В этом случае необходимо знать названия простейших одновалентных остатков, получающихся при отрыве водорода от соответствующих аренов
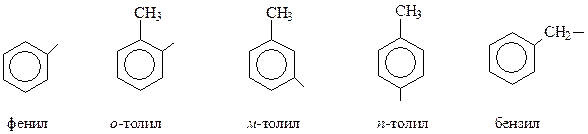
Способы получения
Известно большое число химических реакций, позволяющих получить ароматические углеводороды исходя из других классов органических соединений. Те из них, которые основаны на превращениях соединений алифатического ряда, были уже рассмотрены. К ним относятся: дегидроциклизация алканов (Зелинский, Казанский, Платэ), тримеризация ацетилена (Бертло, Зелинский), циклическая кротоновая конденсация метилкетонов, дегидрирование гидроароматических углеводородов, диспропорционирование циклогексена, циклогексадиена и их гомологов (необратимый катализ Зелинского) и др. Число методов получения аренов может быть существенно расширено за счет модификации известных реакций, а также за счет специальных реакций.
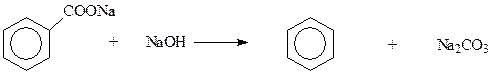
Декарбоксилирование солей ароматических карбоновых кислот при их нагревании со щелочью приводит к аренам
Оказалось, что метод создания С-С-связей по Вюрцу реакцией галогенпроизводных с щелочными металлами успешно может быть применен для синтеза гомологов бензола. Реакция в этом случае называется реакцией Вюрца-Фиттига ( 1864 г.)
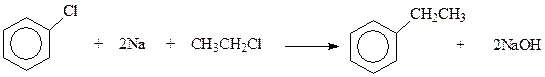
Известно, что необходимость использования в данной реакции смеси двух разных галогенпроизводных ограничивает синтетические возможности реакции, поскольку в результате реакции получается смесь из трех соединений. Это приводит к усложнению лабораторной процедуры и напрасному расходованию исходных материалов. Однако в случае реакции Вюрца-Фиттига дело несколько упрощается, поскольку продукты реакции легко разделяются из-за значительного различия в свойствах. Так, при использовании реакции Вюрца-Фиттига с целью получения этилбензола приходится исходить из хлорбензола и этилбензола. В результате реакции получаются этилбензол (жидкость), бифенил (твердое тело) и бутан (газ), которые легко разделяются. Вместе с тем необходимо отметить, что реакция Вюрца-Фиттига имеет лишь ограниченное применение в синтезе аренов.
Среди всех способов получения аренов особое место занимает алкилирование бензольного кольца в ароматических соединениях. Эта реакция на примере бензола и хлористого метила была впервые осуществлена еще в 1877 году Фриделем и Крафтсом
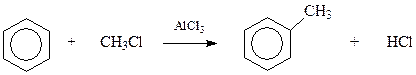
Значение реакции Фриделя-Крафтса в ее универсальности и возможности использования в промышленных процессах. В последующем будет рассмотрен механизм этой реакции как частного случая реакций электрофильного замещения в ароматическом ряду. Пока же обобщим феноменологические наблюдения над реакцией.
Способностью алкилироваться обладают многие ароматические соединения, но не все. Легко вступают в эту реакцию сам бензол и соединения, несущие в кольце так называемые электронодонорные заместители, в том числе и алкильные остатки. По этой причине реакция Фриделя-Крафтса сопровождается полиалкилированием, т.к. продукты первоначального алкилирования обладают большей реакционной способностью, чем исходный бензол. Попутно отметим, что последующая алкильная группа вступает в бензольное кольцо в о - и п -положение по отношению к имеющемуся заместителю
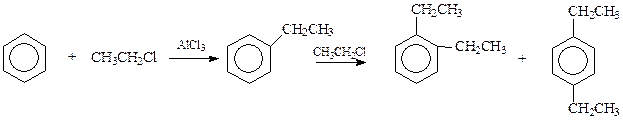
Препятствовать полиалкилированию в какой-то мере можно, используя в реакции избыток бензола.
Выше отмечалось, что в условиях реакции Фриделя-Крафтса легко алкилируются ароматические соединения, несущие в бензольном кольце электронодонорные заместители. Следует оговориться, что это не всегда так. Например, в случае анилина и замещенных аминов не удается успешно алкилировать бензольное кольцо, т.к. аминная группа как сильное основание образует с кислотой Льюиса комплекс и связывает катализатор.
Ароматические соединения с электроноакцепторными заместителями, например, нитробензол, бензолсульфокислота, в реакцию Фриделя-Крафтса практически не вступают.
Фридель и Крафтс для алкилирования бензола использовали галогеналканы. В последующем было показано, что как алкилирующие средства могут быть использованы не все галогенпроизводные. Не пригодны для этих целей соединения, в которых галоген прочно связан с остальной частью молекулы. Например, хлорбензол, хлористый винил и др.
Кроме галогеналканов, в качестве алкилирующих агентов могут быть использованы алкены и спирты. Однако независимо от природы алкилирующего агента, в результате реакции образуются продукты, у которых алкильный заместитель имеет иное строение, чем в исходном галогеналкане, спирте или алкене. Так, при алкилировании бензола хлористым пропилом, пропиленом или пропиловым спиртом получается не пропилбензол, а изопропилбензол
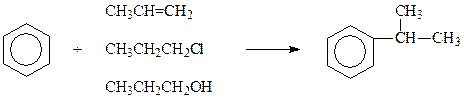
Классическим катализатором реакции Фриделя-Крафтса является хлористый алюминий. Однако реакцию катализируют и другие кислоты Льюиса. Чаще всего применяются трехфтористый бор, фтористоводородная и фосфорная кислоты.
Одним из способов получения аренов является трансформация уже имеющейся в ароматическом соединении боковой цепи в алкильную группу. Особенно часто этот подход осуществляется в отношении жирноароматических кетонов, легко получаемых ацилированием аренов. Благодаря особенностям строения такие кетоны способны восстанавливаться в арены по методу Клемменсена или Кижнера–Вольфа. В первом из них восстановление ведут амальгамированным цинком и соляной кислотой, во втором – гидразином и сильным основанием
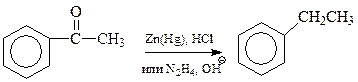
Особенностью рассматриваемого способа получения аренов является то, что в этом случае не происходит перегруппировки боковой цепи и становятся доступными арены с боковыми цепями нормального строения
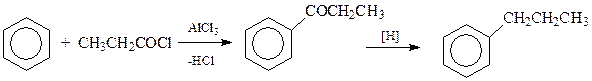
Для получения алкенилзамещенных аренов могут быть использованы все те реакции, которые позволяют получить алкены. В промышленности обычно используется дегидрирование алкилбензолов. Реакция проводится при высоких температурах в присутствии катализаторов дегидрирования – смеси оксидов алюминия, хрома и др.
Наиболее важный представитель алкенилбензолов - стирол получают по следующей схеме, включающей синтез этилбензола по Фриделю-Крафтсу и дегидрирование полученного этилбензола на оксидах хрома и алюминия при 6000C
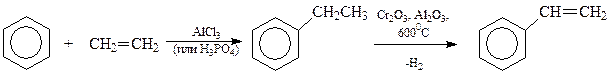
Аналогичная схема используется при промышленном получении еще одного из алкенилбензолов - α -метилстирола
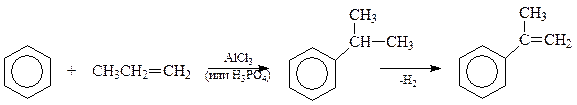
Химические свойства
Молекула аренов включает остаток бензольного кольца и алкильные или алкенильные группы в качестве боковой цепи. Отталкиваясь от строения аренов нетрудно предвидеть основные направления их превращений. Очевидно, что в реакциях с участием бензольного кольца они будут проявлять ароматический характер, вступать в реакции замещения. При обсуждении строения бензола было показано, что он обладает значительной непредельностью. Поэтому в определенных условиях арены могут вступать и в реакции присоединения. Боковая цепь в аренах представляет собой остаток алкана или алкена. В случае реакций с их участием арены будут вступать в реакции свободнорадикального замещения как алканы или же в реакции электрофильного замещения как алкены. Понятно, что реакции по бензольному кольцу будут протекать под влиянием боковой цепи, а реакции с участием боковой цепи будут испытывать воздействие ароматического кольца.
Реакции замещения
11.5.1. Галогенирование. Галогены в зависимости от условий проведения реакции могут реагировать как с ароматическим кольцом, так и с боковой цепью. Каталитическое галогенирование способствует реакции галогена с бензольным кольцом с замещением водородных атомов на галоген. В качестве катализаторов чаще всего используются галогениды металлов, выступающих в качестве акцепторов электронов: FeBr3, AlCl3 и ZnCl2. Роль катализатора сводится к поляризации связи между атомами галогена
Br- δ – Br+ δ --- FeBr3
В результате реакции между ареном и галогеном образуется ароматическое галогенпроизводное. Интересно, что реакция может пойти и дальше, причем второй атом занимает в основном р -положение по отношению к первому
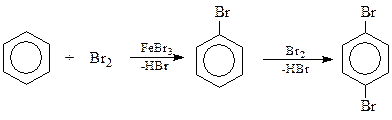
Установлено, что галогены по убыванию реакционной способности в реакции галогенирования аренов располагаются в следующий ряд
F2>Cl2>Br2>I2
Прямое введение атома галогена в ароматическое кольцо практически осуществимо лишь в случае хлора и брома. Фтор в данной реакции слишком активен. Иод, напротив, слишком пассивен. Он переходит в более активную форму (возможно, I+) лишь в присутствии окислителей, например, азотной кислоты
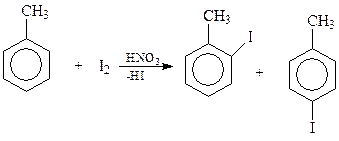
Как и следовало ожидать, под действием ультрафиолетового освещения действие галогена направляется в боковую цепь и реакция сводится к хорошо изученному цепному свободно-радикальному фотохимическому галогенированию. Как и в случае метана, в толуоле последовательно можно заместить на хлор все три водорода метильной группы
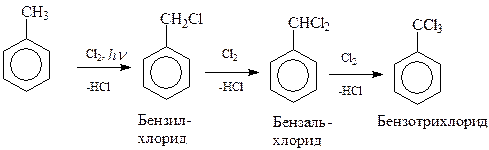
Интересно отметить, что при фотохимическом хлорировании в первую очередь на хлор замещаются водородные атомы ближайшего к бензольному ядру углеродного атома боковой цепи
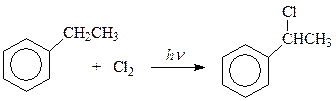
Такое развитие событий связано с образованием в лимитирующей стадии реакции особенно устойчивого радикала бензильного типа
На образование бензильного радикала из толуола требуется лишь 326,59 кДж (для образования трет -бутильного радикала из изобутилена 381,02 кДж, для аллильного радикала 322,40 кДж). Из представленных энергий диссоциаций связей видно, что бензильный радикал содержит меньше энергии и более устойчив, чем даже трет -бутильный радикал. Это объясняет легкость образования бензильного радикала. Устойчивость радикалов бензильного типа связана с участием в делокализации электрона бензольного кольца.
11.5.2. Нитрование. При взаимодействии с азотной кислотой арены замещают свои водородные атомы на нитрогруппу
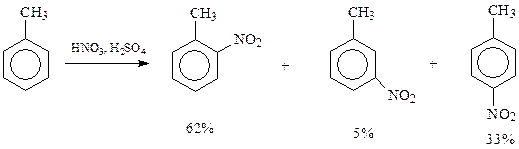
В реакцию нитрования арены вступают легче, чем сам бензол. Благодаря присутствию в толуоле электронодонорной метильной группы удается заместить на нитрогруппу все три способных к замещению водородных атома
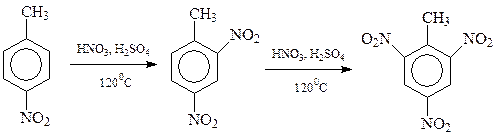
Интересно отметить, что вступившая в кольцо нитрогруппа затрудняет дальнейшее нитрование. Известно, например, что п -нитротолуол в реакции нитрования значительно уступает в реакционной способности толуолу.
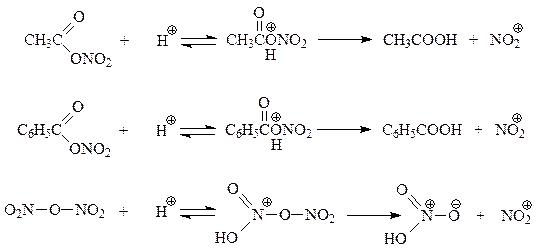
При нитровании реакция идет лучше при использовании в качестве нитрующего агента смеси азотной и серной кислот (1:2). Эта смесь называется нитрующей смесью. Установлено, что в нитрующей смеси происходит реакция между кислотами, в результате которой образуется электрофильная частица – ион-нитрония, которая и участвует в нитровании
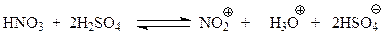
В рассматриваемом кислотно-основном равновесии серная кислота выступает в роли сильной кислоты, а азотная кислота ведет себя как основание. Серная кислота, участвуя в вышеприведенном равновесии, способствует повышению концентрации нитроний-иона в реакционной смеси, что приводит к повышению скорости нитрования. Доказательство роли нитроний-иона при нитровании основано на том, что реакция хорошо идет и с другими источниками этого иона. Так, нитронийфторборат способен быстро нитровать бензол

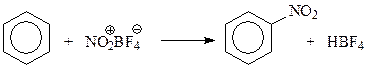
На концентрацию нитроний-иона в нитрующей смеси и на результат нитрования отрицательно влияет вода, которая реагирует с ионом
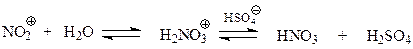
По этой причине при нитровании обычно используются безводные кислоты (дымящие азотная и серная кислоты).
Кроме нитрующей смеси при нитровании могут быть использованы и другие нитрующие агенты. К ним относятся ацетилнитрат, пятиокись азота, бензоилнитрат и др. Ацетилнитрат и бензоилнитрат получают по следующим реакциям
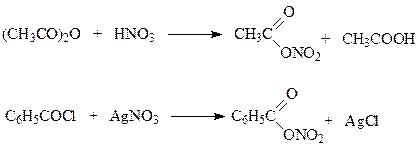
Указанные нитрующие агенты в условиях нитрования генерируют ион-нитрония
11.5.3. Сульфирование. Уже при комнатной температуре при взаимодействии бензола со 100%-ной серной кислотой происходит реакция сульфирования – введения остатка серной кислоты в бензольное ядро. С трудом, но в бензольное кольцо удается ввести и вторую сульфогруппу, которая занимает м -положение по отношению к первой сульфогруппе

Две сульфогруппы настолько дезактивируют кольцо, что ввести в него третью сульфогруппу уже не удается.
При сульфировании образуется также вода, которая разбавляет серную кислоту. Это приводит к снижению скорости сульфирования. К тому же, рассматриваемая реакция является обратимой. С целью предотвращения разбавления серной кислоты и смещения равновесия реакции вправо, сульфирование проводится с удалением реакционной воды по ходу опыта.
Толуол содержит электронодонорную метильную группу, которая способствует сульфированию. Поэтому толуол сульфируется легче, чем бензол и образует смесь о - и п -толуолсульфокислот
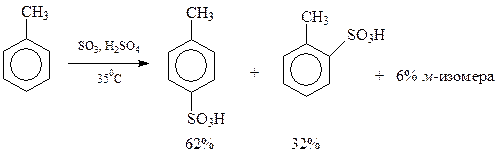
11.5.4. Алкилирование и ацилирование. Алкилирование заключающееся во введении алкильного радикала в бензольное кольцо, уже рассматривалось.
Процесс введения в молекулу органического соединения, в данном случае аренов, кислотного остатка (ацила) называется ацилированием
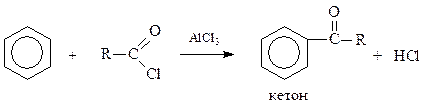
Эта реакция приводит к получению арилзамещенных кетонов.
Поскольку углеводородные остатки активируют бензольное кольцо в реакциях замещения, то все арены достаточно легко вступают в реакцию ацилирования. При этом кислотный остаток занимает о - и п -положение по отношению к алкильному заместителю. Считается, что ароматическое соединение успешно ацилируется, если его реакционная способность не меньше, чем у галогенпроизводных бензола.
Как правило, ацилирование проводится в растворителе, в качестве которого используется сероуглерод, нитробензол или избыток арена.
В качестве ацилирующего агента возможно использование карбоновой кислоты, ее ангидрида или галогенангидрида. Наибольшей активностью обладают галогенацилы, наименьшей – сами кислоты. При ацилировании галогенацилами в качестве катализатора реакции используется хлорид алюминия или другие кислоты Льюиса. В реакции с участием кислот уже требуется использование более сильных катализаторов, например, дымящей серной кислоты.
В ходе реакций алкилирования и ацилирования много общего, они оба относятся к реакции Фриделя-Крафтса. Вместе с тем имеются и различия.
Продуктом ацилирования аренов являются жирно-ароматические кетоны. Оказалось, что они способны образовать комплекс с хлоридом алюминия, который уже не является катализатором
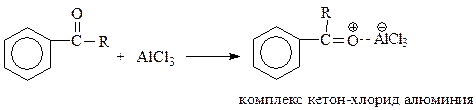
Образование комплекса вызывает ненужное расходование катализатора. К тому же по окончании опыта кетон, являющийся продуктом реакции, приходится выделять разрушением комплекса водой.
Еще одно следствие комплексования кетона с хлоридом алюминия заключается в том, что этот комплекс дезактивирует бензольное кольцо и делает невозможным вхождение в него второй ацильной группы.
Все сказанное остается в силе и при ацилировании в присутствии хлористого алюминия ангидридами кислот. Однако в этом случае ситуация еще более осложняется ввиду того, что и ацилирующий агент комплексуется с молекулой катализатора
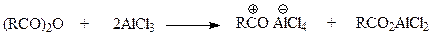
Если образующийся при этом ацилий-катион участвует в реакции, то этого нельзя сказать про комплекс хлористого алюминия с карбоксилатной частью ангидрида. Таким образом, ацилирование арена ангидридом кислоты в присутствии хлористого алюминия протекает таким образом, что один моль хлорида образует комплекс с карбоксилатной частью ангидрида, второй моль дает комплекс с продуктом реакции. В результате в этом случае на моль ацилирующего агента приходится брать более двух молей катализатора.
Реакции присоединения
Среди разнообразных реакций, в которые вступают ароматические соединения с участием бензольного кольца, в первую очередь обращают на себя внимание рассмотренные выше реакции замещения. Это происходит, потому что они протекают вопреки ожиданиям. При той степени ненасыщенности, которая присуща, например, бензолу, этому углеводороду более характерными должны были быть реакции присоединения. При определенных условиях так и происходит, бензол и другие арены присоединяют водородные атомы, галогены, озон и другие способные присоединяться реагенты.
11.5.5. Гидрирование. В присутствии катализаторов гидрирования (платина, палладий, никель) бензол и его гомологи присоединяют водород и превращаются в соответствующие циклогексаны. Так, бензол гидрируется над никелевым катализатором при 100-2000C и 105 атм.:
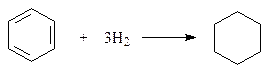
Гидрирование аренов по сравнению с алкенами имеет две особенности. Во-первых, арены значительно уступают алкенам в реакционной способности. Для сравнения с условиями гидрирования бензола укажем, что циклогексен гидрируется в циклогексан уже при 250C и давлении в 1,4 атм. Во-вторых, бензол или не присоединяет, или присоединяет сразу три молекулы водорода. Получить гидрированием бензола продукты частичного гидрирования, такие как циклогексен или циклогексадиен, не удается.
Эти особенности при гидрировании, частном случае реакций присоединения к бензольному кольцу, обусловлены строением бензола. При превращении в циклогексан бензол перестает быть ароматической системой. Циклогексан содержит на 150,73 кДж энергии больше (энергия резонанса) и менее устойчив, чем бензол. Понятно, что перейти в это термодинамически менее устойчивое состояние бензол не склонен. Этим и объясняется меньшая реакционная способность бензола по отношению к водороду по сравнению с алкенами. Присоединение к ароматической системе возможно лишь с участием р -электронов единого электронного облака бензольного кольца. С началом процесса присоединения система перестает быть ароматической и получается богатая энергией и обладающая высокой реакционной способностью частица, которая гораздо охотнее вступает в реакцию присоединения, чем исходный арен.
11.5.6. Галогенирование. Результат взаимодействия галогена с бензолом зависит от экспериментальных условий. Каталитическое галогенирование ведет к образованию продуктов замещения. Оказалось, что ультрафиолет инициирует присоединение атомов галогена к бензольному ядру аренов. Сам бензол на свету присоединяет 6 атомов хлора и превращается в гесахлорциклогексан, представляющий смесь 9 пространственных изомеров
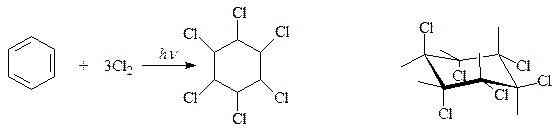
Один из этих изомеров, в котором 3 хлора занимают аксиальные связи, а еще 3 – экваториальные связи (γ-изомер, гексахлоран), оказался эффективным инсектицидом, средством борьбы с вредными насекомыми. Гексахлоран оказался слишком устойчивым в условиях биосферы и способным накапливаться в жировой ткани теплокровных и поэтому в настоящее время не применяется.
По своей реакционной способности по отношению к галогенам в реакциях присоединения бензол значительно уступает алкенам. Например, хлор и бром в четыреххлористом углероде даже в темноте при комнатной температуре присоединяются к циклогексену. В указанных условиях бензол не реагирует. Происходит это только при ультрафиолетовом освещении.
11.5.7. Озонирование. Озонирование - еще один пример, показывающий, что бензол как ненасыщенное соединение может вступить в реакцию присоединения. Озонирование бензола и изучение продуктов гидролиза триозонида было осуществлено еще в 1904 году (Гарриес)
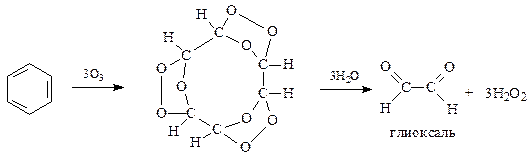
Интересные результаты были получены при озонировании о -ксилола (1941 г., Вибо). Дело в том, что состав продуктов озонирования зависит от положения двойных связей в бензольном кольце. Структура 1 с двойными связями между углеродами бензольного кольца, несущими метильные заместители, при озонировании и гидролизе озонида даст 2 молекулы метилглиоксаля и молекулу глиоксаля
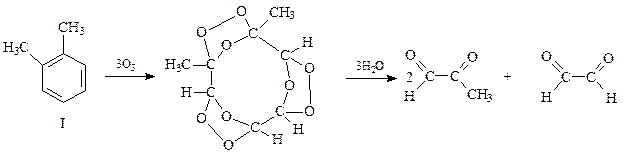
Альтернативная структура II для о -ксилола должна была бы образовать 2 молекулы глиоксаля и молекулу диацетила
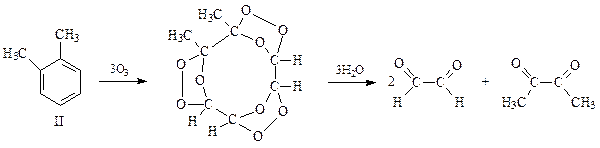
В действительности глиоксаль, метилглиоксаль и диацетил были получены в молекулярном соотношении 3:2:1. Эти результаты свидетельствуют о том, что о -ксилол по строению не I и не II, а как бы некая структура с подвижными, не локализованными двойными связями. Согласно современным представлениям о строении бензола, он не имеет обычных двойных связей и результаты озонирования нельзя рассматривать как доказательство их существования. Эти данные говорят лишь о сильной непредельности бензола и о том, что по чисто формальным признакам ему можно приписать три двойные связи.
Реакции боковой цепи
В аренах в качестве боковых цепей выступают остатки предельных или непредельных углеводородов. Понятно, что в некоторых случаях арены могут участвовать в химических реакциях за счет своих боковых цепей. При этом алкильные заместители придадут арену способность реагировать как алканы, для которых наиболее характерными реакциями являются реакции свободно-радикального замещения. Если в боковой цепи арена находится алкенильная группа, то для него становятся возможными реакции электрофильного присоединения. Бензольное кольцо, сохраняющееся в ходе этих реакций, влияет на скорость и направление соответствующих превращений.
Некоторые из реакций с участием боковой цепи уже рассматривались. Это реакции свободно-радикального хлорирования, восстановления жирно-ароматических кетонов и дегидрирование алкилбензолов. Расширим круг этих реакций новыми примерами.
Окисление аренов. Само бензольное кольцо отличается необыкновенной устойчивостью к действию окислителей. В этом проявляется так называемый ароматический характер. Обычные окислительные реагенты: CrO3, KMnO4, H2O2, OsO4, которые применяются при окислении алкенов, окислить бензол не могут. Конечно, в очень жестких условиях можно окислить и бензольное кольцо. Для этого требуется 450-5000C и наличие катализатора – пятиокиси ванадия. При окислении бензола кислородом воздуха в указанных условиях бензольное кольцо разрушается и получается малеиновый ангидрид
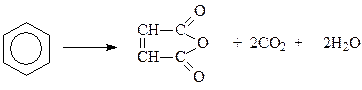
Возможно, это один из лучших технических способов получения малеинового ангидрида.
Все сказанное относительно устойчивости к действию окислителей касается самого бензольного кольца, но не боковых цепей в аренах. Алкильные боковые заместители, в отличие от самих алканов, способны окисляться даже перманганатом калия в водном растворе щелочи.
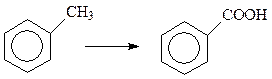
Толуол окисляется уже при 950C количественным выходом в бензойную кислоту.
Интересно отметить, что при окислении аренов действие окислителя всегда направлено на ближайший к бензольному кольцу атом углерода боковой цепи. Поэтому алкилбензолы независимо от длины боковой цепи окисляются в бензойную кислоту
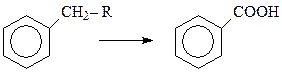
В то же время если этот углерод является четвертичным и не содержит водородного атома, то такой арен в обычных условиях окисления боковой цепи не способен окисляться
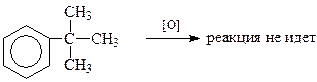
Если бензольное кольцо содержит несколько алкиль

