




Из ранее приведенных примеров окисления углерода ясно, что при низких концентрациях окисляемого элемента наиболее медленным, а, следовательно, "ведущим" звеном процесса может стать массоперенос реагирующих веществ в объеме металлической фазы. Ниже будет показано, что окисление таких обычных примесей чугунов как Si, Mn, P и компонентов природно-легированных чугунов V, Cr и др. до низких остаточных концентраций всегда заканчивается в режиме внутреннего массопереноса. В этом случае скорость процесса окисления определяется интенсивностью поступления атомов растворенного элемента или атомов кислорода в те участки объема металла, где достигаются необходимые для реакции превышения произведений концентраций кислорода и окисляемого элемента над равновесным значением и где существуют другие благоприятные для реакций условия.
В "неподвижном" металле интенсивность перемещения атомов реагирующих элементов определяется величинами их коэффициента диффузии. В интервале температур 1500-1700 °С значения коэффициентов диффузии для кислорода и перечисленных выше элементов по данным большинства исследователей составляют 1×10-4-10×10–4 см2/с. В интенсивно перемешиваемых объемах металла, например, в капле металла, плавящейся в "электромагнитном тигле", т.е. во взвешенном состоянии, эти скорости, естественно, значительно выше. Однако и здесь, в связи с наличием на границах контактирующих друг с другом турбулентных вихревых потоков тонких ламинарных слоев, фактически наблюдаемая скорость массопереноса отдельных элементов тем выше, чем больше скорость молекулярной диффузии для данного элемента (хотя прямой пропорциональной зависимости между ними, разумеется, нет).
При контакте кислорода с расплавленным металлом интенсивность массопереноса в последнем, соответствующая переходу в режим лимитирования внутренний массопереносом, определяется неравенством:  > n
> n  , где – интенсивность поступления кислорода из газовой фазы, отнесенная к единице поверхности металла; – интенсивность поступления к этой поверхности окисляемого элемента; n – величина, характеризующая стехиометрию образующегося окисла элемента "х".
, где – интенсивность поступления кислорода из газовой фазы, отнесенная к единице поверхности металла; – интенсивность поступления к этой поверхности окисляемого элемента; n – величина, характеризующая стехиометрию образующегося окисла элемента "х".
Поток атомов элемента "х" к поверхности металлической капли определяется уравнением:
 , (50)
, (50)
где kx – коэффициент диффузии элемента "х",
F – площадь поверхности металла, контактирующей с окислительным газом;
W – объем металла (капли);
 – число молей элемента "х" в объеме металла V об» W
– число молей элемента "х" в объеме металла V об» W
т.е. 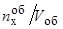 – объемная концентрация элемента "х";
– объемная концентрация элемента "х";
 – число молей элемента "х" в поверхностном слое металла;
– число молей элемента "х" в поверхностном слое металла;
V пов – объем поверхностного слоя, толщина которого определяется приблизительным постоянством концентрации в нем элемента "х", т.е.  – объемная концентрация элемента "х" в поверхностном слое металла.
– объемная концентрация элемента "х" в поверхностном слое металла.
По мере протекания процессов рафинирования металла разность концентраций  постепенно уменьшается и, вследствие приближения поверхностных слоев металла к насыщению их кислородом, доминирующая роль переходит уже к массопереносу кислорода. Многие исследователи-металлурги определяют этот режим как двухстадийное окисление: сначала окисление железа и образование его оксида Fe n O m и далее окисление примесей металла за счет кислорода этого окcида на поверхности капли оксида или в других участках металла, где по тем или иным причинам условия для реакции окисления наиболее благоприятны [27, 30, 45, 47, 51, 58]. Таким образом, режим "внутридиффузионного лимитирования" при очень низких остаточных концентрациях элемента "х" является по существу массопереносом кислорода и примесей металла к этим точкам.
постепенно уменьшается и, вследствие приближения поверхностных слоев металла к насыщению их кислородом, доминирующая роль переходит уже к массопереносу кислорода. Многие исследователи-металлурги определяют этот режим как двухстадийное окисление: сначала окисление железа и образование его оксида Fe n O m и далее окисление примесей металла за счет кислорода этого окcида на поверхности капли оксида или в других участках металла, где по тем или иным причинам условия для реакции окисления наиболее благоприятны [27, 30, 45, 47, 51, 58]. Таким образом, режим "внутридиффузионного лимитирования" при очень низких остаточных концентрациях элемента "х" является по существу массопереносом кислорода и примесей металла к этим точкам.
Сам акт взаимодействия элемента "х" с кислородом завершается образованием зародышей новой неметаллической фазы, на что должна быть затрачена определенная энергия. Величина этой энергии может быть оценена по величинам избыточных над равновесными значениями концентраций элемента "х" и кислорода или, так называемом, переокислении металла – a:
 , (51)
, (51)
где [х]наб и [о]наб - значения наблюдаемых концентраций элементов "х" и "о";
[х]рав и [о]рав – равновесные концентрации этих элементов для данных давлении и температуре:
m и n характеризуют состав образуемого оксида.
Величина кинетического барьера, препятствующего зарождению новой фазы, экспериментально оценивается по значению a, которое весьма различно не только в зависимости от природы окисляемого элемента, но также и от условий зарождения.
Научные основы для изучения условий зарождения новой фазы в объеме до этого гомогенной жидкости положили работы Гиббса, изучавшего кинетику кипения жидкостей [40] и исследования Фольмера [41]. В общем виде скорость образования зародышей новой фазы определяется уравнением:
J = Fexp (–D G /k T), (52)
где F – частотный фактор (число образующихся зародышей новой фазы в секунду);
G – свободная энергия Гиббса (изобарно-изотермический потенциал);
k – константа Больцмана.
Это уравнение применимо во всех случаях, когда в объеме жидкости образуется конденсированная новая фаза (ее собственный кристалл, жидкая капля или кристалл новой фазы продуктов реакций, протекающих в жидкости и т.п.).
При зарождении элементов газообразной фазы, сопровождающемся изменением давления газа в постепенно растущем пузыре, вместо свободной энергии Гиббса используют изохоро-изотермический потенциал: D F * – свободную энергию Гельмгольца.
Тогда:
J = F * exp (–D F */k T). (53)
При протекании реакции, сопровождающейся образованием новой фазы, имеют место процессы:
1. объемный эффект, заключающийся в изменении химического потенциала растворенных в жидкости веществ вследствие протекания реакции и перехода продуктов реакции в новую фазу;
2. поверхностный эффект, заключающийся в затрате энергии на образование в жидкости поверхности раздела и зарождение новой фазы.
Например, в случае образования пузырька СО по реакции
[ C ]+[ O ]={ CO }
имеем:
D F *=4/3(p r 3/ V г)(mг–mр)–4/3pr3 Р г+4pr2gг,м, (54)
где r – радиус образующегося газового зародыша (см);
mг – химический потенциал газообразной оксида углерода (эрг/моль);
mр – химический потенциал растворенной в металле
оксида углерода (эрг/моль), т.е. m[ C ]/m[ O ];
V г – молярный объем газообразной оксида углерода (см3/моль);
Р г – давление, испытываемое газовым пузырьком со стороны внешней среды (дн/см2);
gг,м – удельная свободная энергия на межфазной границемежду газом и металлом (эрг/см2).
Давление:
Р г= Р ат+ Р ф.сг+2gг,м/ r. (55)
Удельную свободную энергию на поверхности металла и инертного по отношению к нему газа обычно характеризуют как "поверхностное натяжение металла" – sг.м (эрг/см2 или Дж/м2).
Различают гомогенное зарождение новой фазы в однородном металле, облечаемое только микроразрывами его сплошности вследствие турбулентного движения металла (кавитационное зарождение новой фазы) и гетерогенное зарождение новой фазы на границе соприкосновения жидкого металла; например, с каплями забрасываемого в его толщу шлака, на межфазной границе металл-огнеупор и др.
Многочисленные попытки расчетов, предпринятые Доброхотовым Н.Н., Андреевым И.А., Богданди Л., Эллиота Дж.Ф и Медоукрофта С.С. [42-45] показали предпочтительность гетерогенного зарождения новой фазы по сравнению с гомогенным. Однако их количественные оценки этого преимущества и скорости процесса зарождения не считаются надежными, т.к. в расчетах принималось допущение о том, что макроскопические свойства жидкости (например, поверхностное натяжение на границе металла и окисляющей фазы, а также металла и образуемого оксида) можно распространять и на микрообъемы зарождающейся новой фазы. Кроме того, во всех исследованиях зарождения новой фазы приходится оценивать химические потенциалы реагирующих веществ по величинам их средних концентраций, т. к. не имеется возможности оценки микронеравномерности их распределения, тогда как для образования элементарных зародышей новой фазы решающую роль, вероятнее всего, играют именно эти макроскопические скопления реагирующих веществ.
С учетом приближенности всех этих расчетов, для характеристики интенсивности гомогенного образования зародышей любой новой фазы вместо самой интенсивности зарождения часто используют пропорциональную ей безразмерную величину – "вероятность зарождения фазы" L, которая определяется уравнением:
 , (56)
, (56)
где V – объем образующейся новой фазы, приходящийся на 1 одну молекулу, V = M /r N
M – молекулярный вес вещества, образующего новую фазу;
r – его плотность;
N – число Авогадро;
sм.з – удельная межфазная анергия на границе металла и зарождающейся новой фазы;
K – константа Больцмана: K= R / N =1,38×10–23 (Дж/°К);
n ¢/ n ∞ – степень пересыщения металла образующимся в нем оксидом, стремящимся к выделению в "новую" фазу.
Например, в случае реакции:
[ Si ]+2[ O ]= SiO 2, n ¢/ n ∞=[ Si ]наб[ O ]наб/[ Si ]рав[ O ]рав (57)
R – универсальная газовая постоянная; R =1,987 (кал/моль×°К) или 8,314 (Дж/кмоль×°К).
Из уравнения (56) видно, что величина вероятности зарождения новой фазы определяется в первую очередь величиной межфазного напряжения sм.з. Эта величина находится в экспоненте изменяется в пределах 40-50 эрг/см2 для кристалла того же химического состава, что и затвердевающий металл и в пределах 900-1200 эрг/см2 для зарождения в расплавленном железе газовых пузырьков (азота или оксида углерода).
Большое значение также имеет и объем, приходящийся на одну молекулу новой фазы. Как видно из уравнения, этот объем обратно пропорционален плотности новой фазы и изменяется от 1-2 кг/м3 для газов до 2000-10000 кг/м3 для конденсированных фаз, встречающихся в металлургии стали. Поэтому как интенсивность, так и вероятность зарождения газообразной фазы из жидкости несравненно меньше, чем, например, большинства неметаллических включений, или кристаллов самого металлического расплава.
Сравнительно малое значение имеет также вероятность зарождения некоторых практически несмачиваемых металлом неметаллических включений, например, таких как Аl 2 O 3, ZrO 2, TiN и др., для которых sм.з по данным различных исследователей составляет от 1600 до 2100 эрг/см2 [43]. Вследствие таких высоких значений sм.з для зарождения образующихся включений, несмотря на их большую плотность, необходимо некоторое пересыщение маточного раствора по сравнению с равновесным, т.е. во всех случаях n ¢/ n ∞>1, следовательно, ln (n ¢/ n ∞)>0. Это положение справедливо как для случая рафинирования металла при кислородной продувке в любом сталеплавильном агрегате, так и при раскислении стали в печах, конвертерах или в сталеразливочных ковшах.
Переокисление металла, обусловленное необходимостью затраты энергии для формирования зародышей новой фазы, было особенно внимательно изучено (Кербер Т. и Эльсен В., Доброхотов Н.Н., Андреев И.А. [44, 45]) применительно к реакции окисления углерода. Это было продиктовано как теоретическим интересом к процессу образования газовой фазы внутри жидкого металла, так и практическими соображениями поиска путей понижения избыточной концентрации кислорода против равновесной с углеродом. Этот вопрос имел огромное практическое значение при доминирующем в то время основном мартеновском процессе производства стали и отсутствии современных методов "внепечной" (ковшевой) обработки металла. Результаты этих исследований сводятся к следующим [45]:
1. Во всех случаях гетерогенное зарождение пузырьков оксида углерода предпочтительнее и протекает более интенсивно по сравнению с гомогенным;
2. Вероятность локальных разрывов сплошности металла и посторонней фазы и, следовательно, влияние контакта металла с этой новой фазой на интенсивность зарождения пузырьков СО и образование газообразной фазы тем больше, чем больше величина межфазного натяжения на границе металла и контактирующей с ним "посторонней фазы" sм.ф., т.е. чем хуже металл ее смачивает;
3. Для температур, обычных при выплавке стали, экспериментально определены величины угла смачивания металлом и адгезии металла к шлакам различного химического состава, к различным неметаллическим включениям и к огнеупорам, образующим "ванну" печей;
4. Наилучшие условия для гетерогенного зарождения пузырьков оксида углерода имеют место при контакте металла с неошлакованной, пористой поверхностью огнеупоров.
5. Значительно менее благоприятна для этого поверхность контакта металла с ошлакованными, относительно легкоплавкими и хорошо смачиваемыми металлом огнеупорами. Еще менее благоприятна для формирования пузырьков СО поверхность контакта металла и гомогенного шлака. Наиболее затруднительно гомогенное образование СО и выделение ее пузырьков.
2.6 Окисление шлакообразующих компонентов
бинарных сплавов с железом
Значительные изменения изобарно-изотермического потенциала и энтальпии системы при окислении растворенных в жидком железе Si или Мn газообразным кислородом обусловливает большие трудности изучения как термодинамики, так и кинетики этих процессов. Поэтому наиболее точные и современные исследования в этой области проведены с использованием в качестве окислительного газа смесей Ar + (3¸82) O 2, CO 2 или Ar + водяной пар (5-10 % Н 2 O) с применением методов плавки во взвешенном состоянии и/или падающей капли (рис. 8).


Окисление кремния было предметом ряда исследований [46-50 и др.] На рис. 8 показано изменение концентрации кремния при различном его исходном содержании (от 1,32 до 5,85;%) при обтекании капли смесью Ar +26,42 CO 2 и исходной температуре 1700 °C Эти опыты показали наличие сходства у процессов окисления кремния и углерода, а именно нулевой порядок реакции[ Si ]+{ O 2} при окислении кремния вплоть до 0,2¸0,3 % и ведущую роль внешнего массопереноса на протяжении этого периода удаления кремния, при таких условиях (т.е. этой исходной температуре и парциальном давлении  ) окисление кремния замедляется после достижения [ Si ]£0,2¸0,5 %; далее оно переходит уже в режим "внутридиффузионного" лимитирования.
) окисление кремния замедляется после достижения [ Si ]£0,2¸0,5 %; далее оно переходит уже в режим "внутридиффузионного" лимитирования.

В работе [46] приведен ряд бесспорных доказательств того, что при высоких исходных концентрациях и упомянутых и t®°С кремний окисляется до SiO, который испаряется с поверхности капли и дает возгоны на более холодных частях аппаратуры. Таким образом здесь, как и в случае окисления углерода, процесс протекает в условиях встречного массопереноса окислительного газа и образующегося газообразного окисла. Однако, как показали исследования [47, 48] такие условия для окисления кремния создаются далеко не всегда. В частности они редко наблюдаются в процессах производства стали.
При относительно небольших начальных концентрациях кремния, при более низких температурах, а, главное, при более высоких парциальных давлениях окисляющего газа происходит относительно быстрое образование кремнеземисто-железистых шлаков. В зависимости от текущих концентраций кремния и от других условий они различаются по химическому составу от фаялитовых (FeO)2× SiO 2 до относительно низко железистых (35-40 % FeO). Окисление кремния при этом переходит в режим лимитирования внутренним массопереносом, преимущественно переносом оксидов железа в шлаковой пленке. В этом случае возможны, а также фактически наблюдаются, два варианта.
1. Активный процесс, когда шлак сохраняется в жидком состоянии, покрывает каплю очень тонким слоем и в большей своей части скатывается и накапливается в нижней части капли. В этом случае процесс протекает относительно быстро.
2. Пассивный процесс, когда образующийся шлак быстро обогащается кремнеземом. Он покрывает поверхность капли и вследствие его высокой вязкости поступление кислорода через его толщу замедляется. В то же время окисление кремния продолжается за счет оксидов железа шлака, он становится все более вязким, т.е. процесс все больше замедляется.
Эти исследования имели чисто научный характер, т.к. условия их проведения очень отличны от наблюдающихся при всех современных процессах производства стали. Наибольший научный интерес, несомненно, представляет определение условий, при которых продуктом окисления являются: газообразная (SiO), жидкие силикаты железа и даже твердая SiO 2.Однако сложность процесса, заключающаяся в одновременном протекании реакций окисления железа и кремния, шлакообразования и испарения SiО не позволила до сих пор выяснить кинетические характеристики каждого из этих совпадающих по времени процессов, например, определить их энергии активации.
Окисление сплавов железа с марганцем исследовано с помощью обоих вариантов метода бестигельной плавки [49, 50] На рис. 9 приведен типичный пример, характеризующий изменение концентрации марганца в условиях обдувания кислородом висящей в электромагнитном поле капли сплава Fе - Мп [48]. Прямолинейное изменение [ Мn ] во времени и незначительная величина энергии активации процесса удаления марганца в области его высоких концентраций (всего 4-4,5 ккал/моль или 16,7-18,8 кДж/моль) свидетельствуют о ведущей роли внешнего массопереноса в этом процессе. Образующиеся шлак, представляющие собой сплавы оксидов железа и оксидов марганца (xFe n O m× yMn k O j) достаточно подвижны даже при содержании марганца в чугуне до 8 % и при температуре металла около 1500 °С и выше; они значительно перегреты над температурой их плавления, т.е. концентрация в них оксидов марганца не превышает 50-60 %. Образующийся шлак не перекрывает верхней полусферы капли, но стекает в ее нижнюю часть, предоставляя таким образом возможность для окисления марганца путем прямого контакта металла капли с кислородом.
Исследование кинетики процесса окисления марганца представляет особые трудности в связи с тем, что одновременно с окислением, сопровождающимся образованием железо-марганцовистого шлака на поверхности жидкого металла, происходит также интенсивное испарение марганца и окисление уже его паров с образованием плотных возгонов в объеме газовой фазы. Специально поставленные опыты с обдуванием аргоном висящей в электромагнитном поле капли сплава Fe - Mn показали, что скорость испарения марганца в нейтральной среде составляет при исходной температуре 1500 °С ~40 % и при 1350 °С ~30 % от общей потери марганца при обдувании капли кислородом при тех же температурах и тех же скоростях течения газа, что и в случае обдувания их аргоном. Таким образом, 60¸70 % марганца от общей его потери успевает окислиться, находясь еще в растворе в расплаве на основе железа и 30¸40 % (в зависимости от температуры и концентрации) марганца успевает испариться и окислиться уже в газовой фазе.
Ведущую роль внешнего массопереноса в процессе окисления марганца подтверждает равномерное микролокальное распределение этого элемента как в "висящей", так и в "падающей капле" (см. рис. 10) [48, 50]. Только при содержании марганца [ Mn ]<4 % в поверхностном слое толщиной менее 0,2 мм наблюдается определенный градиент концентрации марганца. Во всех остальных случаях, за исключением микропор и пузырей, наблюдается равномерное распределение марганца по всему объему капли. Таким образом, даже в объеме неперемешиваемого электромагнитными силами металла (падающая капля) внутренний массоперенос не является ведущим звеном процесса окисления сплавов железа и марганца. Металлографические исследования показали также большое количество мельчайших капелек марганцово-железистого шлака, забрасываемых в металл при его движении на границе контакта со шлаком ("висящая" капля).
В описанных выше опытах с двух-компонентными сплавами Fe - C, Fe - Si и Fe - Mn, а также в опытах с синтетическими сплавами Fe - Cr, Fe - V и Fe - P было установлено, что внешняя массопередача играет важную, в отдельных случаях даже ведущую, роль в процессах окисления металла. При низких концентрациях примесных элементов природа окислительных процессов меняется коренным образом и ведущим звеном дальнейшего протекания процесса окисления становятся уже другие его этапы (например, внутренний массоперенос, адсорбционно-кинетическое звено и т.п.).
 Рисунок 10 – Распределение марганца по диаметру1-граммовых капель марганцовистого чугуна, свободно падающих в кислороде, зафиксированное с помощью электронно-зондового (светлые точки) и лазерного (черные точки) микроанализаторов. Исходная температура образца 14000С. Высота падения капель,см:
1 -30; 2 -60; 3 -90; 4 -120; 5 -150
Рисунок 10 – Распределение марганца по диаметру1-граммовых капель марганцовистого чугуна, свободно падающих в кислороде, зафиксированное с помощью электронно-зондового (светлые точки) и лазерного (черные точки) микроанализаторов. Исходная температура образца 14000С. Высота падения капель,см:
1 -30; 2 -60; 3 -90; 4 -120; 5 -150
|
Поведение фосфора в процессах окислительного рафинирования, как элемента крайне нежелательного для служебных свойств сталей, многократно исследовано. Установлено, что почти всегда окисление фосфора обусловлено образованием прочных фосфатов кальция (СаО)4 P 2 O 5 или (СаО)3 Р 2 О 5. Учитывая ограниченность методов бестигельной плавки, авторы приводят ниже результаты только некоторых исследований первых этапов процесса дефосфорации, не связанных со шлакообразованием за счет специальных присадок флюсующих веществ [48, 49]. Исследования поведения фосфора в чистых сплавах Fе - Р и в чугунах, содержащих [ P ]=1,4¸1,6 %, показали, что вплоть до остаточных концентраций [ P ]=0,30¸0,68 %, в зависимости от температуры металла, его концентрация линейно изменяется со временем (рис. 11), т.е.
– d [ P ]/ d t= const. (58)
 Следовательно, в области высоких концентраций фосфора реакция его окисления имеет нулевой порядок и скорость ее вероятнее всего определяется внешним массопереносом.
Следовательно, в области высоких концентраций фосфора реакция его окисления имеет нулевой порядок и скорость ее вероятнее всего определяется внешним массопереносом.
Продуктом первой стадии окисления фосфора является его газообразный монооксид [59, 60], т.е. процесс окисления фосфора протекает преимущественно по схеме:
[ P ]+1/2{ O 2}={ PO }газ, затем 2{ PO }газ, затем 2{ PO }газ+ +3/2{ О 2}газ=(Р 2 О 5) и (P 2 O 5)+3 Fe ж+3/2{ O 2}газ=((FeO)3× P 2 O 5)
Монооксид фосфора – вещество, устойчивое только в парообразном состоянии при t >1500 °С. Впервые оно было идентифицировано спектрографически [59]. Наибольшая вероятность его образования имеет место именно на контактной поверхности металл-газ т.е. в условиях, когда поступление к этой поверхности атомов фосфора обеспечивается высокой концентрацией этого элемента в расплаве. Изменение изобарно-изотермического потенциала при образовании PO по выше приведенной реакции составляет:
при 1225 °С – 23500 кал/моль (98,23 кДж/моль) и
при 1825 °С – 22000 кал/моль (91,96 кДж/моль), т.е. оно почти не меняется с температурой.


При понижении концентрации фосфора в металле и дальнейшем переходе его окисления в режим лимитирования внутренним массопереносом фосфора и переносом кислорода в объеме жидкого металла, реакция протекает по схеме:
3 Fe ж+8[ О ]+2[ Р ]=(FeO)3× Р 2 О 5 (59)
Данные о термодинамике этой реакции нельзя считать вполне надежными. Однако, в соответствии с результатами А.М.Самарина и др., показавшими что:
D G =1606,9–1,17 T, кДж/моль (60)
и lg K= lg [% P ]2×[% O ]2= –(84200/ T)+31,1 (61)
можно, при условиях конца того или иного сталеплавильного процесса (T =1600 °С и [ O ]£0,05), ожидать, что конечное содержание фосфора будет значительно выше допускаемого ГОСТом, даже если бы в металле присутствовали шлакообразующие, способные растворить (FeO)3 P 2 O 5 и снизить его активность ниже 1, принятой в расчетах [61].
Это приводит к общеизвестному выводу, что окислительное рафинирование от фосфора должно быть основано на образовании фосфатов щелочноземельных или некоторых щелочных металлов. Некоторое представление об извлечении фосфора из металла в условиях его равновесия с основным шлаком дает рисунок 12 [99].

