




Гетерогенными называются химические реакции, происходящие между веществами, находящимися в различных соприкасающихся фазах, т.е. протекающие на поверхности раздела фаз. Гетерогенные реакции являются одной из стадий гетерогенных процессов, хотя часто гетерогенными реакциями называют сами гетерогенные процессы, включающие в себя как одну из стадий химические превращения [24, 25]. В литературе по металлургии этим термином называют многостадийные гетерофазные взаимодействия, протекающие на границах раздела металл-шлак, металл-газ, газ-шлак и т.д.
Главное отличие гетерогенных процессов от гомогенных заключается в том, что они протекают не во всем обьеме системы, а в определенном участке, т.е. на поверхности раздела фаз или в в обьеме одной из фаз гетерогенной системы. В обоих случаях для возможности осуществления реакции необходим подвод реагирующих веществ к участкам системы, где реакция может протекать. К отличительным особенностям всех гетерогенных процессов также относятся их сложность и многостадийность, поскольку здесь сочетаются процессы массопередачи с химической реакцией, что наблюдается всякий раз, когда в контакт приведены две фазы, не находящиеся в химическом равновесии. Такое явление включает в себя ряд элементарных стадий:
1. Массоперенос одного или нескольких реагентов из обьема фазы І к границе раздела фаз. На границе раздела фаз возможно установление физического равновесия во всех случаях, когда концентрации реагентов в обеих фазах вблизи границы конечна.
2. Химическая реакция в фазе 2 или на межфазной границе.
3. Массоперенос реагентов и (или) продуктов реакции в пределах собственно фазы 2, обусловленный градиентом концентрации за счет протекания химической реакции.
Таким образом, кинетика гетерогенных процессов, или макроскопическая кинетика, изучает химическую реакцию в реальных условиях ее макроскопического протекания в природе или в технике, т.е. с учетом побочных физических процессов, накладывающихся на основной химической процесс [26, 27]. В этом ее отличие от классической кинетики, которая изучает протекание химической реакции в идеализированных условиях: при постоянной, как во времени, так и в пространстве, температуре и постоянных в пространстве концентрациях веществ.
Суммарная скорость гетерогенного процесса определяется скоростями отдельных его стадий. Наиболее медленная из перечисленных выше стадий лимитирует процесс, т.е. определяет скорость протекания процесса в целом. Например, если стадия 1). лимитирует процесс, то общая скорость его не зависит от скорости протекания химической реакции и процесс может рассматриваться как явление массопереноса. Сама химическая реакция может быть причиной высокой скорости массопереноса в пределах фазы 2 и поэтому стадия 1). становится лимитирующим звеном всего процесса. Интересен также анализ массопередачи с химической реакцией в том случае, когда процесс лимитируется суммарной скоростью явлений, составляющих стадию 2). Если же скорости отдельных стадий сравнимы между собой, то суммарная скорость реакции не обязательно должна быть равна скорости самой медленной стадии, т.к. все стадии взаимосвязаны.
Таким образом, важнейшими среди физических процессов, накладывающихся на основной химический процесс, является, во-первых, массоперенос исходных веществ и продуктов реакции и, во-вторых, выделение и распространение тепла. Следовательно, макроскопическая кинетика, как наука, представляет собой продукт синтеза двух научных дисциплин: химической кинетики, с одной стороны, и теории процессов массопереноса и теплопередачи – с другой.
Макрокинетика реальных металлургических процессов в ряде случаев определяется процессами переноса. Поэтому очень важно уметь определять характеристики гетерогенного процесса, выявлять лимитирующее звено, количественно описать отдельные стадии или звенья.
В большинстве случаев изучение кинетики гетерогенных реакций сводится, во-первых, к установлению лимитирующего звена и, во-вторых, к определению его характеристик.
Характер режима исследуемого процесса устанавливают с помощью ряда отличительных признаков, главными из которых являются:
1. Зависимость скорости процесса от температуры.
2. Характер кинетической кривой.
3. Экспериментально определяемая величина порядка реакции.
4. Влияние скорости относительного перемещения взаимодействующих компонентов фаз и площади межфазной границы на кинетику процесса.
Выбор того или иного признака определяется конкретной постановкой задачи, а также возможностями и удобствами экспериментального осуществления применительно к данному процессу.
Из сказанного выше следует, что для описания гетерогенных процессов следует применить аппарат формальной кинетики. При этом скорость гетерогенной реакции определяется как количество вещества, реагирующего на единице поверхности за единицу времени и, в отличие от классического определения [4], уже далеко не всегда связана непосредственно с изменением концентрации.
В формальной кинетике для описания зависимости скорости реакции от концентрации участвующих в ней веществ обычно пользуются степенным законом Гульберга-Вааге:
W =k 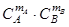 (4)
(4)
где W – скорость реакии;
С А, С В, … – концентрация веществ А, В, …, участвующих в реакции.
Коэффициент пропорциональности k, зависящий для данных веществ А, В и т.д. только от температуры, называется константой скорости реакции или удельной скоростью реакции. Уравнения, подобные (4), выражающие функциональную зависимость между скоростью и концентрацией, называются кинетическими. Сумма показателей степеней, в которых входят концентрации в кинетическое уравнение, называется порядком реакции. При этом различают порядок реакции по отдельному веществу (например, порядок реакции по веществу А есть показатель m A) и суммарный порядок реакции, равный сумме всех показателей:
m = m A+ m B+… (5)
Кинетическое уравнение реакции может быть получено только в результате экспериментального изучения реакции и не может быть выведено из стехиометрического уравнения суммарной реакции.
В том случае, когда скорость реакции не меняется с изменениями концентрации одного или нескольких реагирующих веществ говорят, что эта реакция имеет нулевой порядок. Кинетическое уравнение такой реакции запишется следующим образом:
dC / d t= –k (6)
т.е. константа скорости и скорость реакции равны.
В случае металлургических реакции их порядок удобнее всего определять по концентрации изучаемого компонента в металле А, например, [ C ] и по скорости ее изменения, d [ C ]/ d τ
Скорость реакции первого порядка выражается уравнением:
d [ C ]/ d τ= –k C, (7)
где С – концентрация реагирующего вещества,
τ – время,
d [ C ]/ d τ – скорость изменения концентрации вещества,
k – константа скорости.
Константа скорости реакции первого порядка имеет размерность τ–1 и может быть выражена в обратных секундах (или других единицах времени).
Интегрируя уравнение (7) получим:
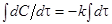 , (8)
, (8)
lnC =–kτ+ S (9)
или
lg [ C ]=(k/2,303)ּτ+ S (10)
где S – постоянная интегрирования.
Таким образом, для реакции первого порядка график зависимости логарифма концентрации реагирующего вещества от времени представляет собой прямую линию.
Если скорость реакции пропорциональна концентрации каждого из двух реагирующих веществ или квадрату концентрации одного из реагентов, то реакция имеет второй порядок. При одинаковых концентрациях реагентов кинетическое уравнение такой реакции имеет вид:
– d [C]/ d τ = kC2 (11)
После разделения переменных и интегрирования получим:
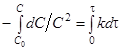 (12)
(12)
и
(1/ C)–1/ C 0=kτ (13)
Здесь " С " или [ С ] – концентрация в металле исследуемого компонента, достигнутая к моменту τ после начала протекания реакции; C o – концентрация того же компонента до начала протекания реакции. Отсюда следует, что для реакции второго порядка должна соблюдаться прямолинейная зависимость между величиной 1/ С и временем.
Реакции, подчиняющиеся простым кинетическим уравнениям, очень редко имеют порядок выше 2-го. Следует иметь в виду, что кинетические уравнения приемлимы лишь к элементарным актам и не описывают суммарную гетерогенную реакцию. Поэтому на основании вида стехиометрического уравнения реакции еще нельзя сделать заключение о действительном механизме реакции. В связи с зтим в кинетику наряду с понятием порядка реакции введено понятие молекулярности, т.е. число частиц, действительно принимающих участие в элементарном акте реакции (в молекулярной – одна частица, в бимолекулярной – две). Таким образом, понятие порядка реакции эмпирическое, а молекулярности – теоретическое. Однако на практике, в особенности при выявлении лимитирующих звеньев гетерогенного процесса, эмпирические кинетические уравнения оказываются чрезвычайно полезными.
Имеется ряд способов определения порядка реакции:
1. Подстановка в формулы. Если реакция имеет нулевой, первый, второй или третий порядок, то в ходе ее должно наблюдаться постоянство константы, полученной подстановкой опытных данных в соответствующее уравнение. Если опытные данные не удовлетворяют ни одному из этих уравнений, значит реакция имеет более сложное кинетическое уравнение, например, дробного порядка.
2. Графические методы. Порядок реакции можно определить, построив ряд графиков, откладывая по одной оси (обычно по ординате) различные функции от концентрации, а по другой – время. В случае реакции нулевого порядка прямая линия получается, если строить зависимость концентрации от времени. Если прямая линия получается в координатах lgC –t, значит реакция в соответствии с уравнением(10) имеет первый порядок. Данные для реакции первого и второго порядков для первых 50 % глубины протекания реакции различаются очень незначительно. В частном случае, когда начальные концентрации всех реагирующих веществ одинаковы, реакция имеет второй порядок, если прямая получается в координатах І / С –t (в соответствии с уравнением (13) и третий порядок, если линейная зависимость имеет место в координатах I / С 2–t.
3. По времени полураспада. В реакциях первого порядка время полураспада (по существу, время, требуемое для превращения любой заданной доли реагирующего вещества) можно определить по кинетическому уравнению следующую частную форму:
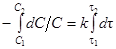 , (14)
, (14)
интегрирование которого дает:
k=(1/t2–t1) ln (C 1/ C 2)=2,303/[ lg (C 1/ C 2)]. 15)
С помощью уравнения (8) можно получить следующее соотношение между временем полураспада и константой скорости:
k=(2,303/t1/2) lg (1/½)=0,693/t1/2, (16)
Таким образом, время полураспада для реакции первого порядка равно:
t1/2=0б693/k, (17)
т.е. в соответствии с уравнением (17) оно не зависит от начальной концентрации. В реакциях второго порядка время, требуемое для превращения половины исходных количеств, в соответствии с уравнением (13) обратно пропорционально начальной концентрации, т.е.
t1/2 = 1/k С о. (18)
Вообще для реакции n-го порядка (кроме 1-го):
t1/2~1/ С n-1. (19)
Подобные соотношения иногда используют для определения порядка реакции.
Итак, характер кинетических уравнений позволяет определить порядок реакции. Это в свою очередь дает возможность, исходя из величины суммарной скорости процесса, вычислить его константу скорости. Температурная зависимость этой величины позволяет определить характер лимитирующего звена процесса.
При повышении температуры и постоянных концентрациях реагентов скорость химической реакции увеличивается вследствие возрастания константы скорости. Ее температурная зависимость выражается уравнением Аррениуса:
dln k/d T = E / RT 2, (20)
где Е – энергия активации. Уравнение (20) подобно уравнению изохоры Вант-Гоффа для температурной зависимости константы равновесия:
dln kc/d T =D U / RT 2, (21)
где D U – изменение внутренней энергии системы.
В формальной кинетике энергия активации определяется как избыток энергии, необходимый для того, чтобы процесс осуществился. Иными словами, для течения реакции необходимо, чтобы исходные вещества преодолели определенный энергетический барьер. После этого они "спускаются" в конечное состояние, т.е. превращаются в продукты реакции. Для определения величины энергии активации необходимо экспериментально найти зависимость константы скорости от температуры. Поскольку
dT / T 2= – d (1/ T), (22)
то уравнение (20) можно представить в виде:
ln k/ d (1/ T)= – E / R. (23)
Отсюда следует, что, если Е не зависит от температуры, должно выполняться соотношение:
ln k= –(E / RT)+ A, (24)
где А – постоянная, т.е. зависимость ln k от (1/ Т) должна быть линейной. Тангенс угла наклона (угловой коэффициент) этой прямой линии в координатах ln k–(1/ T) численно равен E / R.
Из уравнения (20) следует, что когда E ¹ f (T), k= k¥ e–E/RT, где k¥ – константа скорости при бесконечной температуре. Предэкспоненциальный множитель k¥ часто обозначают через А и называют фактором частоты. Чем больше величина Е, тем сильнее зависит k, а следовательно, и скорость процесса, от температуры в соответствии с уравнением Аррениуса:
k= A e–E/RT (25)
Если энергия активации меняется с температурой, то зависимость lg k от 1/ Т будет криволинейной. В этом случае величина энергии активации при данной температуре определяется по наклону касательной к кривой в данной точке.
На практике даже в хорошо поставленных лабораторных опытах далеко не всегда удается создать условия для изучения кинетики самой химической реакции и избежать сопровождающих ее и определяющих скорость всего процесса этапов или стадий. Например, в наиболее близком к идеальному случае окисления газообразным кислородом углерода, растворенного в металлической капле, подвешенной в высокочастотном злектромагнитном поле, реакция:
[ C ]+½{ O 2}={ CO }
Весь процесс возможен только при одновременном протекании таких этапов:
1. Массоперенос кислорода в газовой фазе к поверхности капли;
2. Переход кислорода через границу фаз газ-металл и распространение (массоперенос) его в объеме металла;
3. Сам акт реакции окисления углерода;
4. Образование и выделение из металла в газовую фазу пузырьков оксида углерода.
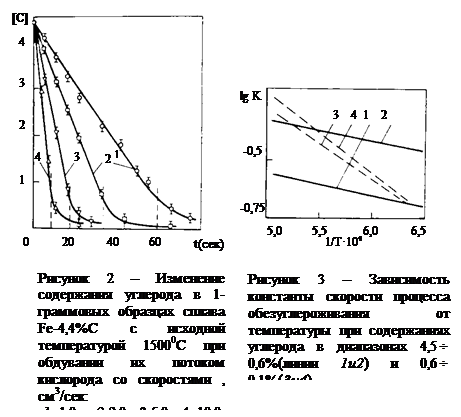
Не вдаваясь пока в подробное рассмотрение всех этих стадий процесса, (рис. 2) отметим лишь, что в определенных условиях каждая из них может оказаться "ведущей". Этим объясняется различные взгляды ученых-металлургов на природу реакции обезуглероживания металла.
Тем не менее, можно указать некоторые признаки для распознавания природы и ведущего звена того или иного процесса. Одним из основных признаков является зависимость скорости протекания процесса от температуры, иными словами, рассчитанная по экспериментальным значениям для разных температур скорость процесса и его энергия активации (см. уравнения (20) или (25)).
Хорошо известно, что скорости химических реакций возрастают с повышением температуры в значительно большей степени, чем скорости многих физических процессов, например, процессов диффузии, или вообще массо- или теплопереноса, адсорбции растворенных в жидкостях веществ в ее поверхностных слоях и т.д.
В интересующем металлургов интервале температур энергия активации химических реакций, в зависимости от природы реагирующих веществ, составляет от 0,01-0,10 до 0,42-1,3 кДж/моль (100-300 ккал/моль) (рис. 3).
Таблица 1 – Изменение химического состава капель
железо-углерод при температурах 1350, 1500 и 1650 °С
| t, с | [ C ], % | |||||
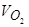 =10 см3/с =10 см3/с
| =5 см3/с
| |||||
| 1350 °С | 1500 °С | 1650 °С | 1350 °С | 1500 °С | 1650 °С | |
| 2,0 | 3,68 | 3,52 | 3,46 | – | – | – |
| 5,0 | 2,51 | 2,25 | 2,05 | 3,44 | 3,32 | 3,17 |
| 7,0 | 1,77 | 1,43 | 1,10 | – | – | – |
| 10,0 | 0,63 | 0,46 | 0,31 | 2,40 | 2,10 | 1,82 |
| 12,0 | 0,40 | 0,22 | 0,10 | – | – | – |
| 15,0 | 0,20 | 0,07 | – | 1,40 | 0,88 | 0,57 |
| 17,0 | 0,12 | – | – | 0,97 | 0,42 | 0,22 |
| 20,0 | 0,06 | – | – | 0,55 | 0,14 | 0,05 |
| 25,0 | – | – | – | 0,18 | 0,03 | – |
| 30,0 | – | – | – | 0,06 | – | – |
В качестве примера определим величину энергии активации процесса обезуглероживания капель сплава Fе - С. На рис. 3 представлены зависимости lg k–1/ T области содержаний углерода выше 0,6 % и ниже 0,5 % для случаев обдувания висящей в электромагнитном поле капли потоками кислорода со скоростями 5,0 см3/с (линии І и 3) и 10,0 см 3/с (линии 2 и 4). Величины констант скорости рассчитаны на основании приведенных в таблице 1 данных об изменении химического состава капель при взаимодействии их с кислородом. Тангенс угла наклона (tg a) прямой І на рисунке равен 1142 и, следовательно,
E a= –2,303 tg a=21,8 кДж/моль (26)
Аналогично при обдувании капли кислородом со скоростью 10 см3/с величина энергии активации процесса составляет 4650 кал·моль–1 (І9,466 кДж/моль-1), т.е. она практически не отличается от измеренной при расходе кислорода 5 см3 /с.
В области содержаний углерода ниже 0,5 % величину энергии активации составили 16 ккал/Моль-1 (66,976 кДж/Моль-1) при расходе кислорода 5,0 см3/с и 18,5 ккал/Моль-1 (77,441 кДж/Моль-1) при расходе кислорода 10,0 см3/с, т.е. в 3-4 раза больше, чем при высоких содержаниях углерода.
Приведенные данные свидетельствуют о том, что при окислении углерода происходит изменение природы и ведущего звена этого процесса. При [ C ]>0,3-0,6 % ведущим звеном, вероятнее всего, является массоперенос кислорода в объеме газовой фазы к поверхности жидкого металла. В зависимости от условий проведения опыта, при концентрациях углерода [ C ]=0,3-0,6 % происходит смена ведущего звена и дальнейшее окисление углерода определяется другими звеньями процесса, например, массопереносом углерода или кислорода в обьеме жидкого металла (капли) к наиболее благоприятным для реакции участкам этого объема.
Как указывалось выше, суммарная скорость гетерогенного процесса определяется скоростями отдельных его звеньев или стадий, причем, если скорость одной из последовательных стадий процесса значительно меньше других, то суммарная скорость определяется скоростью этой наиболее медленной стадии. Если наиболее медленным звеном процесса является подвод реагирующих веществ к зоне реакции или отвод из нее продуктов реакции, то кинетика суммарного процесса будет диффузионной. О таких процессах говорят как об идущих в диффузионной области. В случае, если медленная стадия процесса заключается в химическом или физическом превращении, то скорость процесса определяется скоростью реакции и процесс лежит в кинетической области.
Однако надо помнить, что выводы о характере лимитирующего звена сделаны исходя из положений формальной кинетики и, следовательно, здесь нельзя ожидать абсолютной достоверности, а тем более полного раскрытия природы процесса.
При реакциях между газами и жидкостями или между компонентами флюидных фаз (газов или жидкостей) и твердыми телами, при достаточно малых скоростях потока в движущейся фазе лимитирующим звеном в некоторых случаях оказывается внешняя масоопередача (диффузия). Увеличение скоростей внешних потоков приводит к тому, что скорость гетерогенного процесса начинает определяться уже условиями внутренней массопередачи. Это наиболее типичный случай, если малоподвижной фазой является вязкая жидкость или твердое тело [24, 35, 37]. Признаками того, что процесс определяется условиями внешней массопередачи является:
1. Скорость протекания процесса зависит от скорости потока и природы текущих газа или жидкости, поскольку эти факторы определяют величину так называемого коэффициента массопередачи (6), который представляет собой отношение диффузионного потока к градиенту концентрации и, в зависимости от того в каких единицах они выражены, имеет размерность моль/см2, см/с или 1/с.
2. Диффузионное сопротивление не зависит от времени, из чего следует, что скорость практически также постоянна во времени.
3. Как отмечает А.А.Жуховицкий [24], если процесс прерывается и после некоторого перерыва вновь продолжается в тех же условиях, то его кинетика будет характеризоваться теми же параметрами, что и до перерыва, иными словами, у процесса отсутствует явление "памяти".
4. Скорость процесса сравнительно мало зависит от температуры: энергия активации диффузии газа в смеси [35] приблизительно определяется величиной RT, т.е. для температур сталеплавильного производства (1500-2000 °С) она составляет 2-5 ккал/моль (9-22 кДж/моль), а энергия активации диффузии компонентов в жидких расплавах 10-40 ккал/моль (40-180 кДж/моль).
Если процесс протекает во внутридиффузионном режиме (в высоковязкой жидкости или в твердом теле), то он характеризуется следующими особенностями:
1. Скорость процесса не зависит от скорости движения газа.
2. Скорость существенно зависит от интенсивности перемешивания жидкой фазы или пористости твердой фазы.
3. Диффузионное сопротивление растет со временем, в результате чего количество накопленного или израсходованного вещества пропорционально  [24, 37, 38].
[24, 37, 38].
4. Скорость процесса в значительно большей степени,чем при внешнедиффузионном режиме, зависит от температуры; энергия активации диффузии компонентов в жидких расплавах составляет 10-40 ккал/моль (40-180 кДж/моль) и еще более для диффузии в твердых телах.
Отличительными особенностями гетерогенных процессов, протекающих в кинетической области, являются следующие:
1. Скорость реакции не зависит от скорости газового или жидкого потока.
2. Скорость реакции сравнительно быстро увеличивается с ростом температуры, т.к. в большинстве случаев энергия активации химических реакций существенно больше, чем процессов диффузии (>50 ккал/моль или >200 кДж/моль).

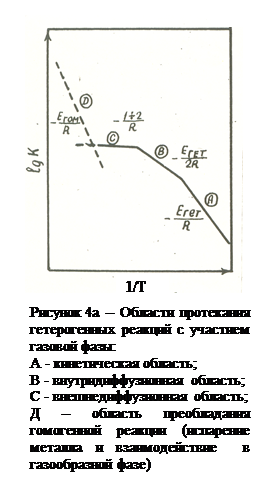
Итак, возможны три области протекания гетерогенной реакции, которые графически показаны в аррениусовых координатах на рис.4а. Как видно из этого рисунка, вследствие больших значений энергии активации, при высоких температурах скорости химических реакций намного больше, чем скорости массопередачи и процессы определяются протеканием диффузии как наиболее медленной стадии. При низких температурах, наоборот, развитие процесса обычно лимитируется скоростью химического превращения.

