




Одними з найбільш важливих і точних методів лабораторних корозійних досліджень є електрохімічні. Найчастіше досліджується зміна потенціалу металу у певному корозійному середовищі в залежності від часу. Через відносно велику тривалість досліджень ця залежність реєструється звичайно за допомогою автоматичного самописа. Більш повну картину корозійного процесу дають так звані поляризаційні криві, за якими судять про поляризуємість даного металу, про роль катодних і анодних реакцій і вплив внутрішніх і зовнішніх факторів на корозійний процес. Особливо важливе місце займають поляризаційні виміри при дослідженні систем, що пасивуються [17,23,33,34].
За поляризаційними кривими можна обчислити швидкість корозії металу. Якщо відома залежність швидкості катодної й анодної реакцій від потенціалу, то шляхом графічної побудови можна визначити швидкість корозії металу в даних умовах. Для того щоб установити залежність між потенціалом і швидкістю електродних реакцій, густина зовнішнього струму повинна бути значно вище густини корозійного струму. Цієї умови необхідно дотримуватись через вплив на величину потенціалу супутнього електродного процесу. Фактично, при знятті катодної поляризаційної кривої швидкість катодного процесу підвищується пропорційно густині зовнішнього струму, але вплив анодного процесу продовжується доти, поки густина зовнішнього струму не стане сумірною з густиною корозійного струму. Графічна залежність, що дозволяє визначити швидкість корозії, представлена на рисунку 5.1. По осі абсцис нанесені значення густини струму в логарифмічних одиницях, а по осі ординат — відповідні величини потенціалу. За кінетичними рівняннями швидкості електродних процесів, залежність між потенціалом і струмом є напівлогарифмічною. На цій підставі побудова поляризаційних кривих у такому масштабі дає можливість шляхом екстраполяції прямолінійних ділянок (які називаються тафелевими) до їхнього взаємного перетинання визначити густину корозійного струму. Оцінка буде правильною, якщо величина стаціонарного корозійного потенціалу не змінюється в часі. Так, наприклад, на рисунку 5.1 показане практичне застосування методу побудови поляризаційних кривих при визначенні швидкості корозії заліза в розведеній соляній кислоті та при додаванні інгібітору корозії [23,33,34].
Цей метод більш складно застосовувати в тих випадках, коли концентраційні зміни в розчині впливають на хід поляризаційних кривих, а також коли на металевій поверхні утворюються фазові й адсорбційні шари. Напівлогарифмічна залежність між потенціалом і густиною струму в цьому випадку порушується і метод стає непридатним [23].
Інгібітор — це хімічна речовина, при додаванні якої в невеликих кількостях у дане корозійне середовище значно зменшується швидкість корозії металів, що знаходяться в контакті з цим середовищем. Як ефективний засіб захисту металів від корозії, застосування інгібіторів набуло особливого значення в останні 40 років у нафтовидобувній, нафтопереробній і хімічній промисловості. Інгібітори широко використовуються для захисту від руйнувань зовнішніх і внутрішніх поверхонь труб і апаратів у циркуляційних охолод-жувальних системах, реакторах для переробки й ємкостях для збереження хімічних продуктів, комунікаційних системах і ін. Їхня велика перевага полягає в тому, що вони придатні при захисті уражених корозією систем без заміни матеріалу або конструкції. Число неорганічних і органічних речовин, які можуть застосовуватися як інгібітори, безупинно збільшується. Але число технічно, екологічно та економічно прийнятних інгібіторів дуже обмежено.
У залежності від способу дії інгібітори бувають плівкоутворювальні (пасиватори) і такі, що адсорбуються (включаючи леткі інгібітори). Інакше їх можна класифікувати як речовини (органічні і неорганічні), що помітно зменшують швидкість корозії за рахунок гальмування анодного процесу (анодні сповільнювачі: NaNО2, K2Cr2O7 ), катодного процесу (катодні сповільнювачі: похідні N, P, O, S - вмісних органічних сполук) або за рахунок впливу на обидва процеси (інгібітори змішаної дії) [23,29-34].
У якості пасиваторів можуть бути використані всі ті речовини, що утворюють з іонами металів нерозчинні продукти і формують плівку. Визначальну роль у їх інгібуючій здатності грає величина рН розчину.
Деякі пасиватори утворюють оксидну плівку на металі. Ця плівка має товщину до 0,01 мкм і може бути в 30—100 разів товстіше плівки, утвореної на поверхні металу під дією повітря. Так, хромати створюють оксидну плівку на залізі, алюмінії і цинку. Однак при несприятливих умовах або малих концентраціях пасиватори здатні прискорювати корозійні процеси. До пасиваторів відносяться кисень, гідроксильні іони, нітрат-, нітрит, фосфат-, молібдат-, хромат-, перманганат-, бензоат-іони та ін. Вони безпосередньо або у вигляді продуктів реакції блокують анодні і катодні ділянки поверхні металу, підвищуючи її потенціал. Їхня велика спорідненість до металу поєднується з високою енергією активації утворення речовин з новими ґратками (хемосорбція). Так, наприклад, кисень при хемосорбції пасивує поверхню, у той час як іони хлору через дуже низьку енергію активації утворення хлоридів металу витісняють адсорбовані на поверхні металу атоми пасиватору.
Фосфати, силікати і бензоати лужних металів є анодними пасиваторами. Так, Na3P04 утворює на анодних ділянках фосфат заліза, що інгібує корозію заліза, зануреного у водний розчин хлориду натрію.
Катіони пасиватору можуть утворювати нерозчинний гідроксид на катодних ділянках кородуючого металу. Якщо занурити залізну пластину в морську воду, що містить MgCl2 на катодних ділянках утвориться плівка з Mg(OH)2; отже, MgCl2 служить катодним інгібітором корозії заліза в розчині хлориду натрію. Це пояснює те, що морська вода менш корозійно-активна для заліза, ніж 3%-ний розчин хлориду натрію.
Деякі речовини мають одночасно анодну і катодну дію. До них відносяться атмосферні пасиватори, наприклад Са(НСО3)2. Іон  утворить карбонат заліза на анодних ділянках, а іон Са2+ — плівку із Са(ОН)2 на катодних ділянках.
утворить карбонат заліза на анодних ділянках, а іон Са2+ — плівку із Са(ОН)2 на катодних ділянках.
Активна концентрація різних пассиваторів знаходиться в межах від 10-5
до 10-1 н. і зростає в такій послідовності: молібдати (10-5 н.), нітрити, вольфрамати, хромати, ацетати, бензоати, силікати, ортофосфати, карбонати (10-1 н.).
При високій концентрації кисень теж діє як пассиватор; наприклад, високолеговані сталі пасивні в аерованих розчинах. Для підвищення ефективності неокислювальних пасиваторів необхідний кисень.
Бензоати, вольфрамати, молібдати, силікати і фосфати не мають окислювальної властивості і пасивують металеву поверхню тільки в присутності кисню; при цьому утворюються оксидні, фосфатні і т. і. плівки.
Бензоат натрію (1%-ний) зменшує швидкість корозії в морській воді і цілком захищає сталі у водопровідній воді, значно зміщуючи потенціал вуглецевих сталей убік позитивних значень. Його пасивуюча дія не виявляється на сірому чавуні, тому що спостерігається ефективна дія гальванопари графіт - ферит.
Ефективна дія гексаметафосфата, ортофосфата, пірофосфата і триполіфосфата натрію й інших поліфосфатів, особливо кальцію і магнію, робить їх цінними добавками до води у водних циркуляційних системах. На поверхні сталі вони утворюють тонкий захисний шар фосфату, на який не впливають зміни температури при рН вище 5. Поліфосфати на відміну від хроматів не небезпечні для здоров'я при низьких концентраціях, але менш ефективні при тих самих концентраціях.
Дія органічних інгібіторів характеризується головним чином їхньою адсорбцією. Відомі три типи адсорбції — фізична, специфічна і хімічна: 1) фізична адсорбція обумовлена існуванням вандерваальсових сил між інгібітором і металом, причому десорбцію легко здійснити промиванням або протиранням, підвищенням температури; 2) специфічна адсорбція супровод-жується блокуванням активних центрів поверхні металу при відносно малих заповненнях поверхні; 3) хімічна адсорбція, або хемосорбція, виникає в результаті відомої хімічної спорідненості між металом і інгібітором, що приводить іноді до утворення на поверхні металу хімічних сполук.
Серед інгібіторів цього типу зустрічаються і неорганічні інгібітори, але основні їхні представники — це органічні речовини, у молекулах яких утримуються певні функціональні групи: С = О, NH, NH2, SH, OH і т. і. Це аміни і їх похідні, етаноламіни, альдегіди, спирти, карбаміди, меркаптани й ін.
Відомо багато органічних сполук, що мають інгібуючу дію. Важливо з'ясувати зв'язок між їхніми захисними властивостями і хімічною структурою. Вивчення захисних властивостей різних органічних сполук переважно в аерованих розчинах дозволило зробити наступні висновки:
а) інгібітори для травлення: сіркувмісні сполуки, похідні ацетилену, слабкі органічні азотвмісні основи, ефективні в кислих розчинах;
б) дія низькомолекулярних сильнолужні амінів (органічних основ) залежить від концентрації гідроксильних іонів;
в) органічні похідні гідроксиламіну відновлюють розчинений кисень, зменшуючи його концентрацію;
г) низькомолекулярні органічні кислоти викликають корозію. Їхні натрієві солі мають захисну дію за умови, що константа дисоціації кислоти має величину порядку 10-4—10-8. При недостатній концентрації інгібітору спостерігається рівномірна корозія;
д) при етерифікації карбоксильної групи кислот їх захисна дія зменшується;
е) феноли мають інгібуючу дію тільки у вигляді фенолятів за умови, що їхня константа дисоціації вище, ніж 10-8, виключення становить о -нітрофенол, що проявляє високу захисну дію;
ж) нітрогрупи надають слабку захисну дію; її можна підсилити шляхом введення в молекулу інших функціональних груп з високою протикорозійною активністю;
з) захисна дія нерозчинних у воді амінів з довгим ланцюгом і їхніх похідних з кінцевою полярною групою пояснюється гідрофобними властивостями цих сполук. Аміни з довгим ланцюгом сприяють утворенню захисних плівок. Вони надають ефективну захисну дію, причому в октилдециламіну вона значно сильніше, ніж у циклогексиламіну;
і) натрієві солі дикарбонових кислот надають таку ж дію, як і нітрит натрію, дихромат калію або дициклогексиламілнітрит;
к) бензоат натрію відомий як ефективний інгібітор. У концентрації 1—2% він надійно захищає сталь, а у водних розчинах блокує дію іонів хлору. У сполученні з нітритом натрію бензоат натрію захищає системи з етиленглико-левими антифризами.
При просоченні пакувального паперу 1%-ним розчином бензоата натрію досягається також надійна захисна дія. Бензоат натрію додають у лакофарбові матеріали й в олії самостійно або в сполученні з нітритом натрію в співвідно-шенні 10:1. В усіх названих випадках він ефективний при рН нижче 6. Таким чином, леткі інгібітори - це органічні або неорганічні, рідкі або тверді речовини з малим, але достатнім для забезпечення адсорбції тиском парів з інгібуючою здатністю. Знаходячись в експлуатаційному середовищі, вони виділяють пари, що контактують з металом, який захищають. Оскільки леткі інгібітори діють на відстані й у газовій фазі, вони викликають величезний інтерес. Значна частина з цих речовин N-вмісні сполуки: аміни іміни, аміак, а також солі амонію (нітрити, карбонати). Їхня дія починається відразу після випаровування. Пари інгібітору розчиняються в тонкому водному шарі, що утворюється на поверхні металу навіть у відносно сухій атмосфері. Насичена інгібітором плівка адсорбується на поверхні металу і створює бар'єр між металом і корозійним середовищем, тобто механізм дії цих інгібіторів є теж адсорбційним.
Отже за механізмом дії – пасивацією або адсорбцією – інгібітори можна віднести до двох груп. Існує і змішаний механізм, що поєднує пасивацію й адсорбцію.
Пасивуюча добавка, що міститься в розчині, буває небезпечною, бо діє як деполяризатор, полегшуючи протікання катодного процесу. Ця речовина повинна бути окислювачем і швидко відновлюватися; такими властивостями володіють іони хроматів, нітритів, молібдатів і вольфраматів. Незважаючи на те що сульфат-, перхлорат- і нітрат-іони є окислювачами, вони повільно відновлю-ються і не можуть служити пасиваторами. Швидкість відновлення особливо важлива в початковий момент контактування пасиватора з поверхнею металу.
Оптимальне інгібування досягається в тому випадку, коли концентрація речовини, що пасивує, стає вище відомої граничної величини. Нижче цієї критичної концентрації речовина поводиться як активний деполяризатор, і швидкість корозії в деяких місцях збільшується — спостерігається точечна корозія. При дуже низьких концентраціях катодна поляризаційна крива перетинає анодну в активній зоні. Тому необхідно, щоб у будь-якій частині інгібованої системи концентрація інгібітору була вище критичної. При інгібуванні корозії заліза за допомогою  ,
,  або
або  критична концентрація становить 10-3 – 10-4 моль/л.
критична концентрація становить 10-3 – 10-4 моль/л.
Випадкові локальні відхилення в концентрації викликають появу точечної корозії, а в присутності деяких іонів (Сl- або  при пасивації заліза) пасивна плівка руйнується.
при пасивації заліза) пасивна плівка руйнується.
Дослідження інгібування заліза хроматами показують, що пасивація пов'язана з утворенням плівки з оксидів заліза і хрому, що містять адсорбовані хромат-іони. Однак більшість агентів, що пасивують, сприяють адсорбції кисню.
Інгібітори-пасиватори зміщують величину корозійного потенціалу в позитив-ний бік. Є інгібітори, що мало впливають на корозійний потенціал, інші – зміщують jс в негативний бік. Кількість речовини, що адсорбується залежить від концентрації інгібітора в середовищі. Установлено, що вище визначеної концентрації речовина адсорбується гірше. Звичайно адсорбційна плівка є мономолекулярною.
Багато авторів вважають, що молекули інгібіторів адсорбуються на поверхні металу, утворюючи захисну плівку. Захисна дія плівки тим ефективніше, чим більше розмір інгібуючої молекули і площа ділянки поверхні, що покривається нею. Якщо молекули інгібітору полярні, то захисний ефект настає при більш низьких концентраціях, ніж при неполярних інгібіторах.
При розчиненні інгібіторів (солей) утворюються позитивно заряджені іони внаслідок дисоціації хлоридів, іодидів тощо або протонування (приєднання Н+ до молекули інгібітора). Активні метали (Zn, Fe, Cd, Ni та ін.) іонізуються при розчиненні, а на поверхні металу накопичується надлишок негативних зарядів. Негативно заряджений метал притягує позитивні іони інгібітору, що адсорбується на металевій поверхні, утворюючи захисну плівку.
Механізм утворення й ефективність захисної плівки підпорядковуються законам адсорбції. Плівка утворюється як на анодних ділянках, так і на катодних за умови, що час від часу швидкість адсорбції на анодній поверхні стає більш високою, ніж на катодній. Незважаючи на пористість (яка може досягати 60%), ця плівка значно сповільнює дифузію іонів і розчинення [23,29-34].
При застосуванні суміші композицій інгібіторів може спостерігатися підвищення або зниження сумарного ефекту. Дуже рідко сумарна активність дорівнює сумі парціальних активностей. Задача полягає в пошуку інгібіторів, суміші яких дають синергізм дії, тобто захисний ефект gS > g1 + g2 + gn + (n-1).
Експериментальна частина
5.3.1 Вказівки до роботи на потенціостаті [22,33]
1. Підготовка зразків. Досліджуваний електрод не повинен мати на поверхні сліди корозії, оксиди. Для цього електрод необхідно зачистити тонким наждаковим папером, повертаючи його навколо осі на 90° так, щоб риски від абразивних зерен на поверхні електроду розташовувалися перпендикулярно, а потім полірують на верстаті з горизонтальним полірувальним колом або вручну на твердій горизонтальній поверхні. Для полірування використовують послідовно не вживану шліфувальну шкурку № 60-80 до М-28 і М-20.
Після полірування електрод знежирюють розчинником (ацетон або ін. очищені органічні розчинники), потім віденським вапном суміш Са(ОН)2, MgО, СаС03 з добавкою 1-2% NaOH. При знежиренні віденським вапном на годинникове скло поміщають невелику його кількість і додають декілька краплин дистильованої води до одержання однорідної кашкоподібної маси, якою за допомогою чистого фільтрувального паперу протирають електрод. Після цього електрод добре промивають дистильованою водою.
При необхідності електрод протравлюють протягом 2-3 хв.: Fe, Ni, Cd - у HCl (1:1) при 20°C; Cu, Ag - у HNO3 (1:1) при 20°C; Pt, Ir - у HNO3 (1:1) і HCl (1:1) при 80-90°C.
Після травлення електрод добре промивають дистильованою водою і висушують чистим фільтрувальним папером.
2. Підготовка посуду, розчинів, комірки. Посуд і електролітичну комірку миють свіжоприготовленим розчином хромової суміші, водопровідною і дистильо-ваною водою. Перед проведенням вимірів комірку 2-3 рази ополіскують робочим розчином, встановлюють робочий електрод (РЕ) і заповнюють робочим розчином.
Для видалення з розчину розчиненого в ньому кисню через комірку протягом 20-30 хв. пропускають очищений водень (інертний газ – N2, He та ін.). Недостатнє очищення робочого розчину приводить до одержання більш низьких струмів обміну і коефіцієнтів переносу.
Для відновлення оксидів, що могли утворитися на поверхні електроду при його підготовці, РЕ поляризують катодно при густині струму 10-4 - 10-2 А/см2 протягом 10-15 хв. Після цього електрод відключають від поляризуючого ланцюга, витримують без струму до встановлення стаціонарного потенціалу і знімають поляризаційну криву.
В комірці повинні бути відсутні гумові пробки і з'єднання. Варто також звернути увагу на положення капіляру Лугіна, що не повинен торкатися електроду, але мусить знаходитися на мінімальній відстані від його поверхні (0,1-0,5 мм). Це положення має бути жорстко зафіксовано.
Використовують електроди порівняння: ртутно-сульфатний закисний (для сірчанокислих розчинів), хлоридсрібний і каломельний (для хлоридних розчинів), окисно-ртутний (для лужних розчинів). Електрод порівняння (ЕП) має знаходитися в розчині на глибині 10-15 мм.
3. Вказівки до порядку роботи на потенціостаті.
1. Прогрів потенціостата 20-30 хв.
2. Тумблер ставиться л положення "+", (якщо РЕ стосовно ЕП заряджений позитивно), якщо негативно - у положення "-".
3. При стаціонарному потенціалі РЕ величина струму, що реєструється міліамперметром повинна дорівнювати 0. Якщо струм реєструється, то при даному початковому потенціалі приймають цю величину струму за вихідну.
4. Електрод витримують без пропущення струму 15 хв., через кожні
3-5 хв., заміряють потенціал електрода до встановлення стаціонарного потенціалу.
5. Потенціал змінюють, починаючи від стаціонарного значення кроком у 5-10 мВ у катодний бік, а після зсуву потенціалу на 100 мВ від стаціонарного - кроком 50 мВ; при кожному потенціалі електрод витримують 2-3 хв.
6. Записують для кожного потенціалу значення струму за міліамперметром в момент включення струму і після витримки в 2-3 хв.
7. Максимальний зсув потенціалу РЕ від початкового повинен бути таким, щоб струм, що протікає через комірку не перевищував 100-150 мА (катодні криві) і 50 мА (анодні криві). Інтервал густини струму звичайно складає від
10-4 до 10-2 А/см2 (повинен змінюватися на 3-5 порядків). Особливо ретельно досліджується область високих густин струму (від 10-4 до 10-2 А/см2).
8. Криві знімають прямим і зворотним ходом, не менше двох разів на свіжо підготовлених електродах. За усередненими даними будують графіки і проводять розрахунки.
Після зняття катодної кривої витримують електрод при φс протягом
10-15 хв. і знімають анодну поляризаційну криву.
5.3.2 Порядок виконання роботи і вказівки до розрахунків за поляризаційними кривими [34].
1. Поляризаційні криві знімають у інгібованих і неінгібованих розчинах: 1н., 0,1 н. і 0,01 н. НСl на сталі 20 (за вказівкою викладача).
2.. Будують поляризаційні криві в напівлогарифмічних координатах φі=f(lgі). φі відкладають за водневою шкалою. У подальшому всі показники знаходять шляхом графічних побудов з поляризаційних кривих (рисунок 5.1) та відповідних розрахунків.
3. За точкою перетину початкових лінійних відрізків (тафелевих ділянок) цих кривих визначають швидкість електрохімічної корозії (саморозчинення) металу - ic і ic’ i (у неінгібованих і інгібованих - „ ’ ” середовищах). Їй відповідає стаціонарний потенціал корозії jс, jс’.
4. При потенціалах  = - 0,20...-0,15 В та
= - 0,20...-0,15 В та 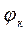 = -0,4…-0,55 В знаходять значення швидкостей парціальних процесів анодної (ia) і катодної (iк) реакції - у неінгібованих і інгібованих розчинах (ia, iк, iа ’, i к’ ).
= -0,4…-0,55 В знаходять значення швидкостей парціальних процесів анодної (ia) і катодної (iк) реакції - у неінгібованих і інгібованих розчинах (ia, iк, iа ’, i к’ ).
5. Визначають тафелеві постійні: ак, аа, вк, ва (як це показано на рисунку 5.1)
 - струм обміну за іонами Н + при
- струм обміну за іонами Н + при 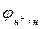 =-0,059pН (при pH=1
=-0,059pН (при pH=1

 -0,06В) - за перетином тафелевих катодних прямих з паралельною вісі абсцис (при
-0,06В) - за перетином тафелевих катодних прямих з паралельною вісі абсцис (при  ).
).
 - струм обміну за іонами Ме Z+ при
- струм обміну за іонами Ме Z+ при 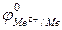 за перетином анодних тафелевих прямих з прямою, паралельною вісі абсцис (за відповідним потенціалом
за перетином анодних тафелевих прямих з прямою, паралельною вісі абсцис (за відповідним потенціалом  )
)

Рисунок 5.1 – Поляризаційні криві корозії сталі у 0,1н. HCl (без Ін та з Ін – “ ’ ”) та схема відповідних побудов та розрахунків
Наближена оцінка рівноважного потенціалу  з урахуванням швидкості ic (точно визначити не можна, тому що j не є оборотними) проводиться за рівнянням Нернста:
з урахуванням швидкості ic (точно визначити не можна, тому що j не є оборотними) проводиться за рівнянням Нернста:
= 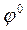 +
+  (5.1)
(5.1)
З урахуванням використовуваного у комірці обсягу розчину кислоти, часу протікання струму, за формулою 5.1 одержуємо:

Встановивши  і знаючи
і знаючи 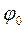 , екстраполяцією (за перетином анодних тафелевих прямих з ординатою при lgi = 0 (шкала в А/см
, екстраполяцією (за перетином анодних тафелевих прямих з ординатою при lgi = 0 (шкала в А/см  ) визначаємо тафелеву константу аа:
) визначаємо тафелеву константу аа:
аа =  , (5.2)
, (5.2)
чим більше аа, тим ефективніше Iн.
Знаючи jк,екстр. За перетином катодних тафелевих прямих з ординатою при lg і =0 (шкала А/см2 ) і  , визначаємо тафелеву константу ак:
, визначаємо тафелеву константу ак:
ак = 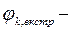 (5.3)
(5.3)
Одержавши значення ак, „-” не враховуємо, вважаючи, що чим більше ак, тим ефективніше Ін.
вк =tg a (a - кут "нахилу" екстрапольованої катодної прямої до осі "х"; ва - те ж, але до анодної екстрапольованої прямої).
Іншими словами вк і ва визначають за похідними:

 (5.4)
(5.4)
6.Розраховують коефіцієнти гальмування: швидкості електрохімічної корозії gс, анодної реакції іонізації металу g а, катодної реакції виділення водню gк , відповідно:
 g a =
g a = 
 (5.5)
(5.5)
7. Визначають  і
і  також із розрахунку:
також із розрахунку:
i = 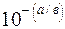 ; =
; =  ; =
; =  (5.6)
(5.6)
Порівнюють зі значеннями, отриманими за поляризаційними кривими.
8. iх наближено визначають за аналітичними вимірюваннями (потенціо-метрія, полярографія, фотометрія – розчинення Me при jс,  =-0,4…-0,7В, або за даними гравіметрії: іх = m ic – іс), А/cм2, іс – із поляризаційних кривих
=-0,4…-0,7В, або за даними гравіметрії: іх = m ic – іс), А/cм2, іс – із поляризаційних кривих
9. Розраховують коефіцієнт гальмування хімічної корозії:
 (5.7)
(5.7)
10.Розраховують часткові коефіцієнти інгібування:
1.кінетичні: 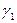 і
і 
-за струмом обміну (позиція 3 – для Fe в кислотах).
- за струмом обміну
У припущенні, що коефіцієнти переносу  і
і  = 0,5 (
= 0,5 (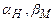 - коефіцієнти переносу для прямого (катодного) і зворотного (анодного) процесів осадження (розчинення) Me;
- коефіцієнти переносу для прямого (катодного) і зворотного (анодного) процесів осадження (розчинення) Me; 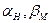 - теж для реакції виділення (іонізації) водню).
- теж для реакції виділення (іонізації) водню).
n- збалансоване число ē, ZМ, ZН – заряди:
| 1 | 2 | 3 | ||
| n=2, Zм=Zн=1 | n=1, Zм=Zн=1 | n=2, Zм=2, Zн=1 | ||
=(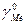 ) ) 
| =() 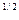
| =() (5.8)
| ||
 2/3 2/3
| 1/2
| 2/3 (5.9)
| ||
 = =  / / 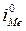 ' ' 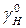 = = 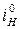 / / 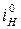 ' (5.10) ' (5.10)
| ||||
 ; ;
| 
| (5.11) | ||
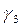 -блокувальний ефект,
-блокувальний ефект,  -ступінь заповнення поверхні Ме інгібітором.
-ступінь заповнення поверхні Ме інгібітором.
 має максимальне значення при великих заповненнях поверхні (тобто найбільш істотний внесок у порівнянні з
має максимальне значення при великих заповненнях поверхні (тобто найбільш істотний внесок у порівнянні з  і
і 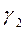 и навіть
и навіть  ).
).
 -енергетичний (подвійношарний або адсорбційний) ефект із ростом
-енергетичний (подвійношарний або адсорбційний) ефект із ростом  або поверхневої концентрації Zм (особливо при полярних або заряжених частках) лінійно зв'язаний з
або поверхневої концентрації Zм (особливо при полярних або заряжених частках) лінійно зв'язаний з  ), збільшується і може перевершувати
), збільшується і може перевершувати 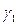 і
і  (навіть при малих
(навіть при малих  ) в 40 і більше разів.
) в 40 і більше разів.
, К=3,3…11, К=3,3 (з урахуванням участі аніонів при достатній концентрації Iн).
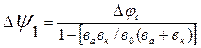 (5.12)
(5.12)
11. Результуючий коефіцієнт гальмування корозії g визначають, як добуток часткових:
g = g1×g2×g3×g4 (5.13) або g = g1+g2+g3+g4 (5.14)
Разом з тим: g = gх×gс (5.15) або g = gх+gс (5.16)
Розраховують g1, g2, g4, а g3 можна розрахувати за (5.13), або (5.14):
g3 = g / g1×g2×g4 (5.17) g3 = g - (g1+g2+g4) (5.18)
Крім того, в розрахуноках враховують долі хімічного та електрохімічного механізму (п. 17), фактично:
g =(Дс ×gс) ×(Д х×gх) (5.19) або g =(Дс ×gс) + (Д х×gх) (5.20)
Оскільки аналітичні вимірювання тривалі, приймаємо gх=10; Д х=0,15 (без Ін) і 0,20 (з Ін)
12. Ступінь інгібіторного захисту:
Z(%)=(1-  )×100% (5.21)
)×100% (5.21)
13.При катодному потенціалі jк = -0,5 чи -0,6 В и анодному потенціалі
j a = -0,1...-0,2 В визначають величини катодних і анодних струмів (iк i i a) при різних рН. Будують графіки залежності lg iк =f(pH) і lg i a = f (pH) і визначають порядки реакцій за H+ ( ) і OH - -іонами (
) і OH - -іонами ( ) за похідними (рисунок 5.2):
) за похідними (рисунок 5.2):
 = n
= n  = m (5.22)
= m (5.22)
14. При струмах iк = i a = 5×10-5…10-3 А/cм2 визначають jк і будують графіки залежності jк = f (pH), ja = f (pH) та визначають значення похідних (рисунок 5.2):
 і
і  (5.23)
(5.23)
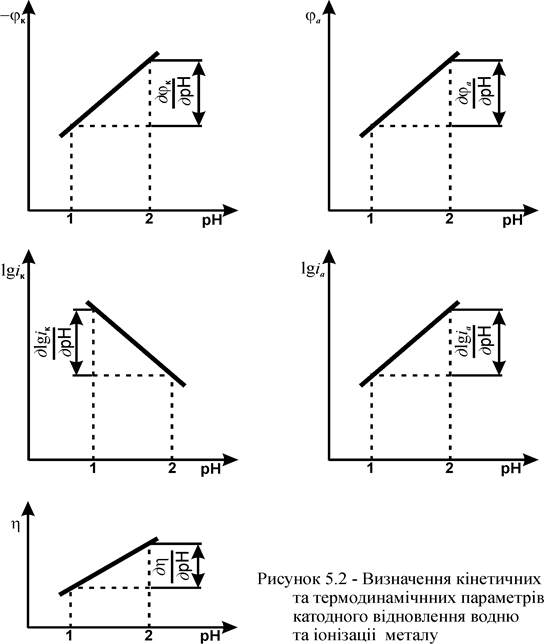
15. Розрахунок значень зазначених вище похідних дає можливість визначити механізм водневої перенапруги катодної реакції, участь у ній тих чи інших іонів.
Наприклад, для деаерованих розчинів:
| Уповільнений розряд | Уповільнена рекомбінація |

| 0,06 |

| 0,5 |

|
16. При швидкості корозії, що дорівнює i c визначають величину катодної (Djк).і анодної (Dja) поляризації (за відхиленням від стаціонарного потенціалу корозії). Потім розраховують ступінь катодного (Cк ) і анодного (C a) контролю за формулами:

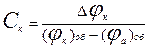 (5.24)
(5.24)
Розрахунок 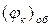 і
і 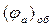 проводять за формулами, наведеними у роботі №3.
проводять за формулами, наведеними у роботі №3.
17. Для одного чи двох розчинів кислоти (за вказівкою викладача) розраховують також частку електрохімічного механізму корозії (Д елх.), для чого використовують дані роботи № 4 (m ic):
 (5.25)
(5.25)
і частку хімічного механізму реакції (Д х ):
 (5.26)
(5.26)
якщо ic = m ic, то механізм корозії – цілком електрохімічний.
18. Водневу перенапругу визначають, як h = ак (при lg i =0, шкала A/см2). а при заданому струмі (10-4...10-1 А/см2) hі визначається як:
 (5.27)
(5.27)
5.4 Висновки: експериментально визначено, кінетичні та термодинамічні параметри корозії сталі, які свідчать про те, що застосовані інгібітори гальмують переважно (...) реакцію, за значенням часткових коефіцієнтів - встановлено, що механізм дії інгібітора – (...); ступінь інгібіторного захисту від корозії становить – (...)
5.5 Тестові запитання для контролю СР
1. Охарактеризуйте переваги та недоліки електрохімічних методів дослідження корозії сталі.
2. Що таке інгібітор корозії?
3. Які переваги інгібіторного захисту ви знаєте?
4. Як класифікують інгібітори?
5. Охарактеризуйте механізм дії пасиваторів.
6. Які типи адсорбції ви знаєте?
7. Охарактеризуйте механізм дії інгібіторрів адсорбційного типу.
8. За яких умов спостерігається оптимальний захист при використанні інгібіторів змішаної дії?
9. Як впливають на корозійний потенціал різні типи інгібіторів?
10. Який порядок роботи на потенціостаті?
11. Як ведеться підготовка посуду, розчинів, комірки для роботи на потенціостаті?
12. Наведіть схему побудов та розрахунків за поляризаційними кривими.
Лабораторна робота №6

