




Синдром Марфана впервые описан в 1896 году французским педиатром А. Б. Марфаном. У больных наблюдается одновременное поражение трех систем: опорно-двигательной, сердечно-сосудистой и органа зрения. Характерными клиническими проявлениями синдрома Марфана являются высокий рост, арахнодактилия (длинные, тонкие, «паукообразные» пальцы рук), гиперподвижность суставов, подвывих хрусталика и миопия, поражение крупных сосудов (аневризма аорты), порок сердца (пролапс митрального клапана. Каждый из этих симптомов может отличаться по степени тяжести и сочетаемости друг с другом у отдельных членов семьи.
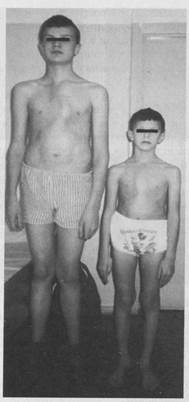
Рисунок 3. Родные братья с синдромом Марфана.
Для синдрома Марфана характерны выраженная плейотропия, варьирующая экспрессивность и высокая пенетрантность. Диагноз синдрома Марфана, ставится при наличии минимум пяти симптомов - аневризма аорты, вывих хрусталика, арахнодактилия, деформация грудины, кифосколиоз. При этом имеет место увеличение (в два раза и более) выведения с мочой глюкозоаминогликанов и их фракций. Особенно резко возрастает почечная экскреция хондроитин-4-6-сульфатов и в меньшей степени - гиалуроновой кислоты и гепаран-сульфата. В моче больных определяется также повышенное содержание (в два и более раз) аминокислоты оксипролина.
Распространенность заболевания в разных популяциях варьирует в пределах от 1:5 до 1:25 тысяч населения. Причиной развития классических форм заболевания 1 типа являются гетерозиготные мутации в гене фибриллина 1 - корового гликопротеина микрофибрилл эластических волокон внеклеточного матрикса, выполняющего в большинстве соединительных тканей архитектурные функции. Ген фибриллина 1 (FBN1) картирован в области 15q21.1, и в настоящее время в нем идентифицировано более 550 мутаций. Эти мутации обладают широким спектром клинических проявлений от изолированной эктопии хрусталика с мягкими скелетными проявлениями марфаноподобного типа до тяжелых неонатальных форм синдрома Марфана, заканчивающихся летальным исходом в течение первых двух лет жизни. При этом мутации, ассоциированные с тяжелыми формами синдрома, локализованы в определенных экзонах гена FBN1. Молекулярно-генетическая диагностика синдрома Марфана как в пренатальном, так и в постнатальном периоде принципиально возможна, но осложняется тем обстоятельством, что мутации в гене фибриллина уникальны, то есть описаны у больных только одной или значительно реже двух или нескольких неродственных семей. Подавляющее большинство из них приводят к классическим формам синдрома Марфана. Однако описаны другие аллельные варианты синдрома, выделяемые клиницистами в самостоятельные нозологические формы -табл. 1.
Важную роль в патогенезе синдрома Марфана играет трансформирующий фактор роста бета (Tgf|3), латентная форма которого непосредственно связана с фибриллином 1. Предполагается, что при снижении уровня фибриллина 1 повышается активность Tgf|3, следствием чего является высвобождение протеаз, участвующих в медленной деградации эластических волокон и других компонентов внеклеточного матрикса. Однако эти предположения требуют дополнительного экспериментального подтверждения.
Наряду с синдромом Марфана и его аллельными вариантами, известны другие марфаноподобные аутосомно-доминантные заболевания, не связанные с мутациями в гене FBN1. Так, мутации в гене FBN2, кодирующем изоформу 2 фибриллина, найдены у больных синдромом Билса - арахнодактилия, контрактуры суставов кистей рук, ки-фосколиоз, аномальная форма ушных раковин, генерализованная остео-пения, которая может способствовать развитию деформаций различных отделов опорно-двигательного аппарата. Для данного заболевания патология сердечно-сосудистой системы и органа зрения не характерны. При редком атипичном варианте синдрома Марфана найдены мутации в одном из генов коллагена I типа (COL1A2). Марфаноподобный фенотип наблюдается также при прогрессирующей диафизарной дисплазии 1 типа Камурати-Энгельманна, вызванной мутациями в гене Tgfpi - TGFBL Второй тип синдрома Марфана связан с мутациями в гене рецептора 2 Tgfpi - TGFBR2. Мутации в гене TGFBR1 найдены у больных синдромом Фурлонга - марфаноподобной болезнью II типа, сочетающейся с краниосиностозом и умственной отсталостью. Аллельным вариантом каждого из этих двух заболеваний является синдром аневризмы аорты Лоеса-Диетза. Этот синдром часто путают с синдромом Марфана, так как оба заболевания имеют перекрывающиеся спектры клинических проявлений. В таблице 1 представлены варианты наследственных скелетных дисплазии с марфаноподобным фенотипом.
Таблица 1.
Некоторые аллельные варианты синдрома Марфана и наследственные скелетные дисплазии с марфаноподобным фенотипом
| Нозологическая форма | Основные клинические критерии диагностики | Ген, первичный биохимический дефект |
| Синдром Марфана, тип 1 | дилатация или расслоение аорты, вывих/подвывих хрусталика, тяжёлая миопия, скелетные аномалии - высокий рост, долихостеномелия, арахнодактилия, деформация грудины, сколиоз, кифоз, дуральная эктазия и др. | FBN1, фибриллин 1, коровый компонент микрофибрилл эластических волокон |
| Марфаноидный скелетный синдром | марфаноидный фенотип без сердечно-сосудистых и глазных аномалии | |
| Эктопия хрусталика, семейная | эктопия хрусталика, мягкие скелет- ные проявления, отсутствие кардио-васкулярной патологии | |
| MASS-синдром (Mitral valve, Aorta, Skeleton Skin) | пролапс митрального клапана, рас- ширение корня аорты, скелетные аномалии, истончение, атрофические полосы (стрии) кожи, ранняя миопия | |
| Синдром Марфана в сочетании с синдромом Шпринтцен-Голдберга | фенотипические проявления синдрома Марфана в сочетании с краниосиностозом, скафоцефалией, гипотонией, эндофтальмом, гипе- рэластичностью кожи, ректальным диастазом, вертикальной таранной костью и умственной отсталостью | |
| Синдром Вейла-Марчезани, аутосомно-доминантный | низкий рост, брахидактилия, туго- подвижность суставов и аномалии хрусталика |
Наследственные факоматозы
Факоматозы характеризуются сочетанным поражением нервной системы, кожных покровов и внутренних органов. Это название впервые предложил в 1923 году голландский офтальмолог J. van der Hoeve, описавший опухолевидные невоидные образования на сетчатке глаз при ту-берозном склерозе. Термин происходит от греческого слова "факон" - невус. Для многих факоматозов характерна варьирующая экспрессивность. Наряду с тяжелыми клиническими формами, отличающимися крайне неблагоприятным прогнозом, существуют стертые и олигосимптомные варианты. Наследуются факоматозы преимущественно по аутосомно-доминантному типу с неполной пенетрантностью, достигающей во многих случаях 75-90%. Факоматозы подразделяются на две большие группы: бластоматозы (нейрофиброматоз, туберозный склероз) и ангио-матозы (цереброретинальный ангиоматоз - синдром Гиппеля-Линдау, атаксия-телеангиэктазия, энцефалотригеминальный ангиоматоз и др.). Все факоматозы обусловлены мутациями в генах, относящихся к классу супрессоров опухолей.
Нейрофиброматоз I типа или болезнь Реклингхаузена-Уотсона -наиболее распространенная форма факоматоза с частотой среди населения 1:2500-3000. Клинически нейрофиброматоз I типа характеризуется тетрадой симптомов, описанных еще в 1930 F. J. Darier. (1) На коже туловища и конечностей больных наблюдаются разнокалиберные "кофейные" пятна, в динамике имеющих тенденцию к нарастанию по количеству и размеру. Кожные изменения могут также проявляться в виде участков депигментации, телеангиэктазий, гипертрихоза-рис.56. (2) Характерными клиническими проявлениями заболевания являются доброкачественные опухоли кожи и подкожной клетчатки - нейрофибромы, состоящие из смеси клеток Шванна и фибробластов. (3) Часто наблюдаются опухоли нервных стволов и окончаний, значительно варьирующие по форме, величине, количеству. (4) Для многих больных характерно отставание в физическом и умственном развитии различной степени выраженности.
Ген NF1 характеризуется необычайно высокой частотой возникновения мутаций. Более 50% пациентов имеют вновь возникшие мутации, причем в подавляющем проценте спорадических случаев (90%) мутации имеют отцовское происхождение. Наиболее вероятным механизмом этой мутационной нестабильности, выражающейся в форме геномного импринтинга, является нарушения процесса метилирования гена NF1. Белок, дефектный при нейрофиброматозе I типа получил название нейрофибромин. Он активно экспрессируется в эндотелии сосудов и в гладко-мышечных клетках. В 80% случаев мутации в гене NF1 приводят к преждевременному окончанию синтеза белка. Большая часть из них представлена протяженными внутригенными перестройками. Соматические мутации в гене NF1 идентифицированы в опухолевых клетках злокачественных меланом, нейробластом, анапластических астроцитом, спорадических карцином кишечника и других тканей.
Нейрофиброматоз II- типа встречается с частотой 1 на 33-40 тысяч новорожденных. Болезнь дебютирует, в среднем, в возрасте 21-22 лет. Выделяют центральную и спинальную формы заболевания. Клиническая картина определяется локализацией опухолей в веществе головного и спинного мозга. Опухоли мозга могут сопровождаться повышением внутричерепного давления, в результате которого появляется головная боль, рвота. Очаговая симптоматика зависит от места расположения опухоли и вовлечения в процесс черепно-мозговых нервов. Наиболее часто поражается слуховой нерв: невринома слухового нерва может быть одно- или двусторонней, причем в последнем случае может отмечаться грубая деформация продолговатого мозга. У больных с центральным нейрофиброматозом II типа, наряду с типичными двусторонними мультифокальными акустическими невриномами (опухолями восьмого краниального нерва, производными от клеток Шванна), могут развиваться менингиомы, билатеральные вестибулярные шваниомы, шваниомы дорзальных корешков спинного мозга и presenile lens opacities. Также как и при нейрофиброматозе I типа частыми являются изменения со стороны костной системы (задержка роста, сколиоз, кифоз, псевдоартроз, локальный гигантизм). У большинства пациентов с нейрофиброматозом II типа кофейные пятна и периферические нейрофибромы либо полностью отсутствуют, либо их число не превышает шести.
Наследование нейрофиброматоза II типа имеет те же особенности, что и при заболевании I типа, хотя и менее выраженные. Геномный им-принтинг выражается в более раннем дебюте и тяжелом течении заболевания при получении мутантного аллеля от матери, по сравнению с теми пациентами, которые унаследовали свою мутацию от отца. Потеря и/или аберрантное состояние локуса NF2 является важным элементом развития не связанных с нейрофиброматозом II спорадических швани-ом и менингиом, которые вместе составляют около 30% всех первичных опухолей мозга. Так что продукт гена NF2 также относится к супрессорам опухолей.
Туберозный склероз, описанный французским неврологом D.-M. Bourneville (1880) и английским дерматологом J. J. Pringle (1890), представляет собой гетерогенную группу аутосомно-доминантных заболеваний с неполной пенетрантностью и варьирующей экспрессивностью. Популяционная частота заболевания различна в разных возрастных группах - 1 на 30 000 среди взрослых и вдвое больше - 1 на 15 000 - среди детей в возрасте до 5 лет.
Туберозный склероз или эпилойя (эпилепсия плюс анойя - умственная отсталость) характеризуется триадой симптомов: а) кожными изменениями б) судорожными припадками; в) психическими расстройствами со значительным снижением интеллекта. Клинические проявления заболевания складываются в зависимости от преимущественного поражения головного мозга (в виде разрастания глии и появления атипичных муль-типолярных клеток в области бугорков), либо кожных покровов. Для последних характерно сочетание пигментированных пятен с участками депигментации в различных отделах туловища и конечностей. Специфичны изменения кожи, часто наблюдаемые в поясничной области в виде «шагреневой кожи». Возможно проявление фиброзного ангиоматоза в области крыльев носа и подбородка. Типичны ахромичные листовидные пятна, околоногтевые фибромы, аденоматозные разрастания сальных желез в виде «adenoma sebaceum» на спинке носа и в виде «бабочки» на щеках. У большинства больных уже в детском возрасте имеется та или иная степень снижения интеллекта, нарастающая в процессе жизни больного. Умственная отсталость встречается примерно у 70% больных и усугубляется вследствие деструкции мозга. Возможны изменения со стороны глаз в виде застойных сосков или атрофии зрительных нервов, эндокринные нарушения, пороки развития внутренних органов.
Первыми признаками заболевания у детей в возрасте 3-4-х месяцев могут явиться судорожные припадки тонического характера, затем они становятся полиморфными, плохо поддаются лечению. Иногда на глазном дне в области диска зрительного нерва обнаруживаются специфические разрастания, носящие название «тутовая ягода». Для младенческой формы заболевания характерны кардиальные и глазные гамартомы. Неврологические и психические нарушения являются результатом гамар-томных туберозных образований по ходу мозговых оболочек, извилин коры головного мозга. Чаще они появляются в области базальных ганглиев, стенок желудочков мозга, реже в области мозжечка, продолговатого мозга. Гамартомы представляют собой скопления атипичных ги гантских ганглиозных клеток, а в стенках желудочков содержат, кроме того, ангиоматозную ткань. Возможно вовлечение в опухолевый процесс других органов - почек, печени, сердца с последующей склонностью к малигнизации.
Туберозный склероз представляет собой генетически гетерогенную группу аутосомно-доминантных заболеваний со сходной клинической картиной. Примерно 40% семей с туберозным склерозом имеют I тип заболевания, обусловленный мутациями в гене TSC1, локализованном в области 9q32-34. В подавляющем большинстве оставшихся семей болезнь обусловлена мутациями в гене TSC2, расположенным в области 16р13.3. Это II тип туберозного склероза. Белки, кодируемые генами TSC1 и TSC2, были названы гамартином и туберином соответственно. Эти два белка взаимодействуют друг с другом с образованием туберин-гамартинового комплекса, участвующего в негативной регуляции инсу-линового сигнального пути. Гиперэкспрессия в системе in vitro каждого из генов TSC подавляет рост и пролиферацию клеток, а также изменяет их морфологию.
В гене TSC2 обнаружено несколько сотен различных мутаций, значительная часть которых составляют протяженные внутригенные деле-ции. Подавляющее большинство мутаций гене TSC1 приводит к образованию укороченных форм белка - это небольшие делеции, инсерции и нонсенс мутации. Для туберозного склероза характерна высокая частота возникновения новых мутаций, достигающая по разным оценкам 60-65%. В целом, мутации в генах TSC1 и TSC2 объясняют подавляющее большинство семейных и примерно половину спорадических вариантов туберозного склероза с превалирующим вкладом гена TSC2 во втором случае. Клиническая диагностика двух генетических форм заболевания, обусловленных мутациями в генах TSC1 и TSC2, практически невозможна, хотя очевидно, что умственная отсталость значительно чаще встречается у носителей мутаций в гене TSC2. Молекулярная диагностика туберозного склероза достаточно трудоемка из-за генетической гетерогенности заболевания, большого количества экзонов и разнообразия мутаций в каждом из двух генов.

