




P и c.9.2
6. Стандартные растворы.
Раствор № 1. Растворяют 500 мг нефтепродукта в 50 мл ацетона; 1 мл полученного раствора содержит 10 мг нефтепродукта.
Раствор № 2. Разбавляют раствор № 1 в 5 раз ацетоном; 1 мл полученного раствора содержит 2 мг нефтепродукта.
Построение градуировочного графика.
В градуированные пробирки вместимостью 10 мл, снабженные притертыми пробками, помещают 0; 0,1; 0,2;...; 0,7 мл стандартного раствора № 2, разбавляют каждый раствор ацетоном до 2 мл, приливают в каждую пробирку по 8 мл раствора желатина, закрывают пробкой и перемешивают.
Оптические плотности образовавшихся эмульсий измеряют на фотоэлектроколориметре (или фотометре) по отношению к холостой пробе (2 мл ацетона плюс 8 мл раствора желатина) при длине волны λ = 508 нм, пользуясь кюветой с расстоянием между стенками 1 см. Строят график, откладывая по оси ординат оптические плотности, а по оси абцисс - соответствующие им содержания нефтепродуктов, в мг.
Подготовка активного угля и приготовление сорбционной колонки.
Активный уголь обрабатывают 1 ч в приборе Сокслета диэтиловым эфиром, высушивают сначала на воздухе, потом 5-6 ч при 220 °С. После этого уголь дробят и отбирают фракцию 0,5-1,0 мм.
В небольшую стеклянную трубку длиной 10 см и диаметром 1 см, снабженную краном и стеклянной дырчатой пластинкой на дне, помещают стеклянную вату слоем 1 см, всыпают 1,5 г подготовленного активного угля и покрывают тонким слоем стеклянной ваты. После этого сорбент осторожно уплотняют стеклянной палочкой.
Ход определения
Собирают прибор, как показано на рис. 9.2. Сточную воду в объеме 500-1000 мл, содержащую 1-20 мг летучих нефтепродуктов, помещают в склянку Дрекселя и пропускают 2 ч воздух через фильтр при комнатной температуре. Если сточная вода содержит только бензин, отдувку можно проводить 1 ч. При очень малых концентрациях летучих нефтепродуктов в пробе отдувку повторяют, если надо, несколько раз, взяв новые порции воды по 1 л и продувая каждый раз в ту же колонку с активным углем. Водоструйный насос протягивает воздух через воду и колонку с углем со скоростью 2-3 пузырька в 1 с, нефтепродукты поглощаются активным углем. Для каждого определения следует брать свежую порцию угля. Затем разбирают прибор и десорбируют нефтепродукты с угля ацетоном. Сначала вливают в колонку 5 мл ацетона, дают ему пропитать уголь, потом пропускают ацетон через уголь порциями по 2 мл (всего 5-6 порций), собирая элюат в градуированную пробирку с притертой пробкой. Каждую порцию ацетона оставляют на угле 5-10 мин, закрывая в это время колонку притертой пробкой и кран. В пробирку с элюатом вставляют притертую пробку, перемешивают, отбирают ал и квотную часть 2 мл и переносят ее в другую такую же пробирку. Затем, медленно, всегда с одинаковой скоростью, вливают по стенке пробирки 8 мл раствора желатина, закрывают пробкой и перемешивают, переворачивая пробирку. Измеряют оптическую плотность образовавшейся эмульсии по отношению к холостой пробе при длине волны λ =506 нм и по градуировочному графику находят соответствующее содержание летучих нефтепродуктов в мг.
Расчет
Содержание летучих нефтепродуктов в анализируемой сточной воде (X, в мг / л) определяют по формуле:
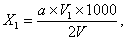 (9.2)
(9.2)
где а - содержание летучих нефтепродуктов, найденное по графику, мг;
V 1- общий объем элюата, мл;
V - объем пробы, взятой на анализ, мл.
9. 1.1.2. Определение нефтепродуктов при концентрации их выше 0,3 мг / л
Аппаратура.
Колонка с сорбентом. В качестве колонки используют трубку длиной 10 см и диаметром 1 см с впаянной в нее дырчатой стеклянной пластинкой; нижний конец трубки оттянут до диаметра 1 мм. В колонку помещают слой (1 см) стеклянной ваты, слой (2-3 см) сорбента (этого количества достаточно для поглощения 50-100 мг полярных соединений) и снова тонкий слой стеклянной ваты.
Реактивы:
1. Неполярные растворители - экстрагенты. Применяют один из следующих растворителей.
Гексан х. ч. (т. кип.68,8 °С, плотность 0,66 г / см 3).
Тетрахлорид углерода х. ч. (т. кип.76,7 °С, плотность 1,59 г / см 3).
Пентан х. ч. (т. кип. 36,07 °С, плотность 0,63 г / см 3).
Петролейный э ф ир х. ч. (т. кип.40-70 °С, плотность 0,63-0,67 г / см 3).
Несмотря на марку х. ч. все перечисленные растворители часто оказываются недостаточно чистыми для данного определения, поэтом у их рекомендуется пропустить через колонку с активным оксидом алюминия, предварительно высушив прокаленным сульфатом натрия.
2. Хлороформ х.ч.
3. Сульфат натрия безводный х. ч. Перед применением прокаливают и охлаждают в эксикаторе.
4. Сорбент. Применяют один из следующих сорбентов.
Оксид алюминия для хроматографии, II степени активности. Продажный препарат этой марки обычно достаточно чист, но вследствие большой гигроскопичности может содержать избыточную воду. Его надо перед применением прокалить 4 ч при 600 °С. Другие препараты могут быть сильно загрязненными. Их надо после измельчения до частиц, размером 0,1 мм обработать в аппарате Сокслета тетрахлоридом углерода 6-8 ч, а потом после испарения из него СС l 4смочить дистиллированной водой, перемешать и прокалить 4 ч при 600 °С.
Силикагель марки КСК, размолотый и просеянный через сито с диаметром отверстий от 0,10 до 0,07 мм и предварительно просушенный 24 ч при 1 20 °С. При сомнении в его чистоте (загрязнение железом) его надо очисти т ь кипячением в разбавленной (1:1) соляной кислоте, промыть дистиллированной водой до удаления хлорид - ионов, высушить и прокалить 24 ч при 120 ° С.
Ход определения (для извлечения органических веществ используется хлороформ).
Остаток после отгонки или отдувки летучих нефтепродуктов, а в тех случаях, когда это возможно (см. выше), непосредственно отобранную пробу сточной воды объемом 100-3000 мл (в зависимости от предполагаемого содержания нефтепродукта) переносят в делительную воронку подходящей вместимости, приб а вляют соляную кислоту плотностью 1,19 г / см 3до значения рН меньше 5, приливают 20 мл хлороформа и сильно встряхивают несколько минут. Затем дают постоять 10-15 мин до разделения слоев, сливают нижний хлороформный слой в колбу с притертой пробкой, стараясь при этом не захватить ни воды, ни промежуточного слоя эмульсии. Водный и промежуточный слой в воронке (или, е сли надо, перенося его в другую делительную воронку) обрабатывают второй порцией хлороформа и полученный экстракт присоединяют к первому экстракту. Двух экстракций обычно бывает достаточно. Затем небольшим количеством хлороформа (около 50 мл) обмывают стенки сосуда, в котором находилась проба до экстракции, переносят в ту же делительную воронку, взбалтывают, оставляют на некоторое время, выливают слой хлороформа в колбу с экстрактом и отгоняют хлороформ. Если для экстракции была взята порция пробы 3 л, то лучше, разделив ее на три части, проводить экстракцию из каждого литра пробы отдельно и все экстракты соединить.
Экстракт высушивают, поместив 1 г безводного сульфата Na в колбу с экстрактом или на фильтр, выпуская через него экстракт из делительной воронки в колбу.
Присоединив колбу с экстрактом к холодильнику, помещают ее на горячую закрытую электроплитку и отгоняют хлороформ, пока в колбе останется 1 0-20 мл хлороформного раствора. Нагревание прекращают, дают колбе остыть и разбирают прибор.
Оста тки хлороформа удаляют при комнатной температуре. Небольшой, предварительно взвешенный бюкс помещают в вытяжном шкафу на расстоянии 25-35 см от комнатного вентилятора. Бюкс наполняют на 3/4 его объема полученным экстрактом и включают вентилятор. По мере испарения хлороформа в бюкс подливают экстракт до тех пор, пока он не будет перенесен полностью. Колбу, где был экстр ак т, обмывают несколькими малыми порциями хлороформа, перенося их в тот же бюкс. Когда в бюксе останется м енее 0,3 мл хлороформа, на что обычно требуется 30-40 мин, выключают вентилятор и продолжают испарение на воздухе, взв ешив ая бюкс, закрытый крышкой, каждые 1-2 мин. Результаты взвешивания наносят на график, соединяют полученные точки и за конечный результат принимают тот, котор ый соответствует точке перелома на образовавшейся кривой.
Разность между массой бюкса с остатком после удаления хло р оформа и массой пустого бюкса показывает общее содержание экстрагируемых хлороформом веществ.
Остаток после отгонки хлороформа обрабатывают 1-2 мл сухого и чистого гексана (или петролейного эфира, кипящего при 40-70 °С) и переносят раствор или суспензию в колонку с оксидом алюминия, под которую подставляют маленькую колбу. Бюкс, в котором был остаток после отгонки хлороформа, несколько раз промывают маленькими порциями гексана, перенося каждую порцию в колонку с окисью алюминия. После этого промывают колонку еще несколькими порциями гексана, собирая их в ту же колбу. Не следует при этом допускать, чтобы уровень гексана в колонке опускался ниже верхней границы слоя оксида алюминия.
Непосредственное экстрагирование гексаном, без предварительной экстракции хлороформом, приводит к заниженным результатам (ошибка может доходить до 30 %), если анализируемая вода содержит взвешенные частицы. На их поверхности наряду с нефтепродуктами сорбируются вещества, растворимые в хлороформе, но не растворимые в гексане (асфальтены, смолы, нафтеновые кислоты и т. п.), препятствующие экстрагированию гексаном нефтепродуктов. Если же анализируемая вода прозрачная и лишена взвешенных частиц, или, если заведомо известно об отсутствии указанных мешающих веществ, то экстракцию можно сразу проводить гексаном или другим неполярным растворителем тем же способом, что и хлороформом, затем отогнать большую часть гексана на водяной бане и пропустить через колонку с оксидом алюминия, как описано выше.
Получив таким образом раствор нефтепродуктов в гексане, освобожденный от примеси полярных соединений, удаляют гексан, испаряя его в бюксе при комнатной температуре с помощью вентилятора так же, как раньше удаляли хлороформ. Когда в бюксе останется примерно 0,5 мл раствора, испарение продолжают без вентилятора, взвешивая бюкс каждые 2 мин. Перед каждым взвешиванием бюкс закрывают крышкой и после взвешивания вновь снимают крышку для дальнейшего испарения. Когда масса бюкса перестанет изменяться, испарение и взвешивание прекращают.
Расчет.
Содержание основной массы нефтепродуктов (малолетучих) в мг / л (Х 2) находят по формуле:
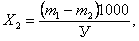 (9.3)
(9.3)
где m 1- масса бюкса с остатком после удаления гексана, мг;
m 2- масса пустого бюкса, мг.
У - объем пробы, взятой для определения, мл.
Суммарное содержание летучих и нелетучих нефтепродуктов равно X 1 + X 2.
Примечание. Разность между общим количеством экстрагируемых хлороформом веществ и количеством нефтепродуктов равна содержанию других экстрагируемых этим растворителем веществ, нерастворимых в гексане и полярных, включая нафтеновые кислоты и фенолы. При желании получить еще более точные результаты анализа, можно выделить и взвесить кислотные вещества (нафтеновые кислоты, фенолы и т. п.) Для этого гексановый раствор перед пропусканием его через оксид алюминия переносят в делительную воронку, фильтруя через очень маленький фильтр, если он получился мутным, и промывают фильтр гексаном. Раствор в делительной воронке обрабатывают 1 м раствором едкой щелочи, разделяют водный и гексановый слои и промывают гексановый слой водой до исчезновения щелочной реакции, присоединяя промывные воды к водному раствору. Гексановый раствор пропускают через ок с ид алюминия и заканчивают определение нефтепродуктов, как описано выше. А водный раствор подкисляют, извлекаю т из него кислотные соединения диэти ло вым эф иром, отгоняют эфир и взвешивают.
При работе, таким образом, результат анализа будет представ л ен тремя значениями:
1)содержанием нефтепродуктов;
2) содержанием нафтеновых кислот и других кислотных соединений;
3)содержанием других веществ, экстрагируемых хлороформом.
9. 1.1. 3. Определение нефтепродуктов при их концентрации ниже 0,3 мг / л
В этом случае 3-3.5 л анализируемой воды недостаточно для получения надежных результатов, а экстрагировать нефтепродукты из еще больших объемов воды, применяя обычную экстракционную аппаратуру, нецелесообразно. Выделить нефтепродукты можно одним из следующих способов.
1. Экстрагировать нефтепродукты из 10-20 л анализируемой воды, применяя од и н из приборов для непрерывной (в потоке) жидко - жидкостной экстракции. Высокий коэффициент распределения нефтепродуктов между органическим растворителем и водой позволяет пропускать через прибор воду с довольно большой скоростью.
2. Пропустить такой же объем анализируемой воды че ре з адсорбент, извлекающий из нее нефтепродукты (активированный уголь, специальный сорбент типа «экоперль»,«перлит» и т. п.), а затем провести десорбцию хлороформом или четыреххлористым углеродом. Ход проведения адсорбции - десорбции при использовании в качестве адсорбента активированного угля следующий. Уголь предварительно освобождают от веществ, изымаемых хлороформом. Для эт ого его обрабатывают в течение некоторого времени хлороформом в приборе Сокслета, завернув в фильтровальную бумагу, затем извлекают из прибора, высушивают на воздухе и прокаливают при температуре темно - красного каления. В бюретку высотой 25 см и диаметром около 1 см насыпают примерно 1,5 г активированного угля (слоем 12-13 см) и пропускают через эту колонку 10-20 л анализируемой воды со скоростью 0,4 л / ч (около 7 мл / мин).
Затем извлекают уголь из бюретки, помещают его на часовое стекло, размещая тонким слоем, и дают ему высохнуть на воздухе до постоянной массы (веса), после чего переносят в гильзу прибора Сокслета (или заворачивают в фильтровальную бумагу) и экстрагируют в этом приборе в течение 24 ч. Хлороформный экстракт обрабатывают, как описано в разделе 9.1.1.2.

