




N -замещенные фенилгидразины 43 реагируют с 1-хлор-4-пентином 44 с образованием гидразонов, которые далее превращаются в триптамины 45 по механизму Грандберга.(Схема 15) [17] Введение заместителей в этиленовый фрагмент триптамина данной реакцией не представляется возможным, по причине неустойчивости замещенных хлорпентинов.
Схема 15.

Существует также метод Шапиро-Абрамовича [18], в котором аминогруппа скрыто защищена в лактамном цикле (схема 16). При этом гидразон (46) образуется по реакции Яппа-Клингесмана между 2-оксопиперидин-3-карбоновой кислотой, генерируемой из соответствующего этилового эфира 47, и катионом диазония, а перегруппировка протекает с образованием оксокарболина 48. Гидролиз амидной связи и декарбоксилирование дают триптамин 49.
Схема 16.

Относительно недавно был разработан метод синтеза триптаминов 50 с помощью тандемной реакции гидроформилирования и синтеза индолов по Фишеру, позволяющий вводить заместители в альфа положение этиленового фрагмента.[19]
Схема 17.

Реакция прохдит с хорошими выходами и может быть проведена стереоселективно, если использовасоть оптически активный алиламин 51. Единственным, но существенным минусом этойспо реакции является применение дорогостоящего катализатора.
Другие способы.
Существует ряд методов, в которых так же, как и в синтезе по Фишеру, образование пиррольного цикла и аминоэтильного фрагмента происходит одновременно. Флеминг и др. [20] разработали метод синтеза триптаминов 52 из диаминоспиртов 53 под действием аммиака в метаноле (схема 18). При этом диаминоспирты синтезировали из аминокетонов 54, которые легко получались присоединением диалкиламинов к арилвинилкетону 55 по Михаэлю.
Схема 18.

Стратегия синтеза триптаминов, предложенная Николау [21], заключается в последовательном превращении защищенных Boc-анилинов 56 в спирогетероциклы 57, а затем в о -замещенные анилины 58, циклизация которых в условиях кислотного катализа приводит к образованию триптаминов. 59 (Схема 19) Причем при использовнии трифторуксусной кислоты происходит снятие защитной группы, в то время как при использовании концентрированной соляной кислоты снятия Boc-группы не происходит.
Схема 19.

Однако вводить заместитель в боковую цепь триптамина с помощью этих методов представляется сложным, так как эти синтезы накладывают большие ограничения на исходные соединения.
Обсуждение результатов
Разработанная ранее в нашей лаборатории циклопропилиминиевая перегруппировка гидразонов циклопропилкетонов 3 открывает путь к синтезу производных 2-(1 H -индол-3-ил)этанаминов (триптаминов) 4 и 1,4,5,6-тетрагидропиридазинов 5, первые из которых обладают ярко выраженной биологической активностью (см. Схема 1). Механизм данной реакции включает раскрытие циклопропанового кольца галогенид-ионом, циклизацию в пирролин и сигматропную перегруппировку, аналогичную синтезу Фишера.
Данная работа посвящена исследованию данной перегруппировки в ряду замещенных в циклопропановом фрагменте гидразонов, генерируемых in situ из соответствующих кетонов и арилгидразина. В качестве модельного реагента был выбран 4-бромфенилгидразин, так как при его реакции с метилциклопропилкетоном (МЦК) триптамин образовывался с наибольшим выходом.
Синтез исходных α-β непредельныхкетонов 60 осуществлялся путем альдольно-кротоновой конденсации ацетона 61 и замещенных бензальдегидов 62. (Схема 20)
Схема 20

Сначала нами был выбран метод, предполагающий использование небольшого количества щелочи (1,75 эквивалентов) и значительный избыток ацетона (10 эквивалентов). Однако скорость реакции существенно снижалась со временем и при использовании описанного протокола реакционная масса содержала значительные количества промежуточного продукта реакции, что осложняло выделение целевого кетона.Применение другого метода, предполагавшего значительное количество щелочи (12 эквивалентов) и всего 4 эквивалента ацетона позволило упростить процедуру выделения целевого кетона.
Циклопропанирование полученных α-β непредельныхкетонов 60 было осуществлено реакцией Кори – Чайковского действием иодида триметилсульфоксония в присутствии щелочи в диметилсульфоксиде. Реакции проходили с хорошими выходами. В чистом виде арилзамещенные циклопропилкетоны 63 были выделены методом колоночной хроматографии.
Схема 22

Основной задачей при исследовании циклопропилиминиевой перегруппировки гидразонов замещенных циклопропилкетоновбыло определение направления реакции и подбор условий для ее региоселективного проведения. Раскрытие циклопропанового фрагмента гидразона, может проходить как по наиболее, так и по наименее замещенной связи, что может привести как к α- так и к β- замещенному этиленовому фрагметну тритапмина (Схема 23)
Схема 23

Для осуществления подбора условий в качестве модельного вещества, взаимодействующего с 4-бромфенилгидразином 64, был выбран 2-фенилциклопилэтанон По аналогии с незамещенными циклопропилкетонами перегруппировка была проведена при кипячении в спирте (4 мл на 0,6 ммоль кетона) в течении 16 часов. После проведения реакции, была проведена экстракция из воды хлороформом. Целевое вещество осталось в водном слое, так как гидрохлориды аминов растворимы в воде и плохо растворяются в хлороформе. Водная фракция была упарена и проанализирована методом спектроскопии ЯМР 1H. Анализ спектра показал присутствие двух продуктов со спиновой системой, соответствующей 5-замещенному индолу с преобладанием одного из них.. Как было выявлено впоследствии, минормный продукт является результатом раскрытия циклопропилгидразона по наименее замещенной связи.
При смене растворителя на ацетонитрил и проведении реакции в таких же условиях (4 мл растворителя на 0.6 ммоль кетона, кипячение в течение 16 часов) реакция проходиит более селективно. Нерастворимый в ацетонитриле гидрохлорид триптамина выпадает в процессе реакции в виде осадка, что позволяет облегчить процесс выделения целевого продукта. Анализ этого осадка с помощью спектроскопии ЯМР 1H показал, что побочный продукт образуется, но в значительно меньших количествах. Разбавление реакционной массы (8 мл ацетонитрила на 0.6 ммоль кетона) позволило избежать образование побочного продукта. Данные условия позволяют получать целевой триптамин без примеси побочных продуктов. Разделение и анализ фильтрата показал, что, как и ранее, помимо триптамина 65 образуется арилзамещенный тетрагидропиридазин 66, причем тоже селективно.
При помощи спектроскопии ЯМР 1H представляется сложным однозначно определить, при каком атоме углерода этиленового фрагмента находится заместитель, то есть по какой связи прошло раскрытие циклопропанового кольца. Но с использованием двумерной спектроскопии COSY установлено, что бензильный протон (4.2 м.д.) взаимодействует с протонами аммонийной группы (8.7 м.д.), на основании чего можно однозначно утверждать, что продуктом исследуемой перегруппировки является триптамин с заместителем в альфа положении этиленового фрагмента. Проведение реакций с большими загрузками и другими ароматическими заместителями в циклопропановом фрагмента кетона, также показало, что во всех случаях раскрытие циклопропанового фрагмента гидразона проходит по наиболее замещенной связи (Таблица 1).
Таблица 1.
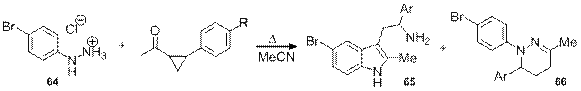
| R | Соединение 65 | Соединение 66 |
| H | 61% | 5% |
| Me | 68% | 9% |
| F | 50% | 19% |
Экспериментальная часть
Спектры ЯМР 1Н и 13С регистрировали на спектрометрах «Brüker AC-200» (200.13 и 50.3 МГц), «Bruker AC-300» (300 и 75.5 МГц соответственно) и «Bruker DRX-500» (500 МГц и 125.3 МГц соответственно) для растворов в CDCl3, (CD3)2SO, содержащих 0.05% Me4Si в качестве внутреннего стандарта; эксперименты COSY, NOESY и HMBC выполнены на спектрометрах «Bruker AVANCE II 300» и «Bruker DRX-500». Масс-спектры регистрировали на приборах «Finnigan MAT INCOS-50» (ЭУ, 70 эВ, прямой ввод) и «Finnigan LCQ» (ESI). Тонкослойную хроматографию проводили на пластинках Silicagel 60 («Merck»). Для препаративных разделений использовали хроматографию на колонке (силикагель 60, 0.040–0.063 мм, «Merck») при соотношении вещество–сорбент, равном ~1: 100. В реакциях использовали стандартные продажные реактивы и химически чистые перегнанные растворители.
3.1. Синтнез α-β непредельных кетонов.
В плоскодонную колбу, оснащенную магнитной мешалкой и содержащую 50мл 10% водного раствора NaOH (120 ммоль) прибавили арилальдегид 1 (10ммоль) и ацетон 2 (40ммоль). Реакционная масса перемешивалась при комнатной температуре 3 часа, после чего продукт реакции 3 был экстрагирован из воды хлористым метиленом. Органический слой осушили сульфатом магния и упарили. Все три реакции проходили при загрузке в 5г и продукт в чистом виде выделялся кристаллизацией из системы петролейный эфир/ этилацетат 3:1.
4-Фенилбут -3-ен-2-он: Выход-81%. Спектр ЯМР 1H (CDCl3): δ = 2.17 (c, 3H), 6.30 (д, J = 12 Гц, 1H,), 7.09 (д, J = 12 Гц, 1H), 7.38–7.90 (м, 5H).
4-(4-Толил)бут -3-ен -2-он: Выход-85%. Спектр ЯМР 1H (CDCl3): δ 7.89 (с, 2H), 7.58 (с, 3H), 6.69 (с, 1H), 2.37 (д, J =16.0 Гц, 6H).
4-(4-Фторфенил)бут-3-ен -2-он: Выход-67%Спектр ЯМР 1H (CDCl3): δ 7.56 – 7.46 (м, 3H), 7.10 (т, J = 8.5 Гц, 2H), 6.65 (д, J = 16.3 Гц, 1H), 2.38 (с, 3H).

