




Генные болезни - это разнородная по клиническому проявлению группа заболеваний, обусловленных мутациями на генном уровне.
Основой для объединения их в одну группу являются этиологическая генетическая характеристика и соответственно закономерности наследования в семьях и популяциях. Поскольку мутации в индивидуальных генах - этиологический фактор генных болезней, то закономерности их наследования соответствуют менделевским правилам расщепления в потомстве, т.е. формальная генетика генных наследственных болезней ничем не отличается от «поведения» в семьях любых менделирующих признаков. Необходимо, однако, сразу сделать пояснения в отношении содержания понятий генных мутаций и менделирующей наследственности у человека.
Во-первых, согласно многочисленным исследованиям разных наследственных болезней и генома человека, в целом можно говорить о многообразии видов мутаций в одном и том же гене, которые являются причиной наследственных болезней. У человека описаны следующие виды генных мутаций, обусловливающие наследственные болезни: миссенс, нонсенс, сдвиг рамки считывания, делеции, вставки (инсерции), нарушения сплайсинга, увеличение числа (экспансия) тринуклеотидных повторов. Любой из этих видов мутаций может вести к наследственным болезням. Даже одна и та же генная болезнь может быть обусловлена разными мутациями.
Во-вторых, современная генетика, принимая в полной мере менделизм, делает некоторые «поправки»: условность понятий о доминантности и рецессивности, неоднородность проявления ал-
леля, унаследованного от отца или матери, - импринтинг, сложный характер взаимодействия генов, гонадный мозаицизм и т.д. Более того, установлено, что мутации в разных частях одного гена ведут к разным болезням. Например, мутации в разных частях гена «RET-онкоген» ведут к четырем клинически разным наследственным болезням: двум формам множественной эндокринной неоплазии (ZA) и (ZB), семейной медуллярной тиреоидной карциноме, семейной болезни Гиршпрунга.
Мутации, вызывающие наследственные болезни, могут затрагивать структурные, транспортные, эмбриональные белки, ферменты.
Существует несколько уровней регуляции синтеза белков: претранскрипционный, транскрипционный, трансляционный. Можно предположить, что на всех этих этапах, осуществляемых соответствующими ферментативными реакциями, могут возникать наследственные аномалии. Если принять, что у человека примерно 80 тыс. генов, а каждый ген может мутировать и обусловливать другое строение белка, то, казалось бы, должно быть не меньшее число наследственных болезней. Более того, по современным данным, в каждом гене может возникать до нескольких сотен вариантов мутаций (разные типы в разных участках гена). На самом деле более чем для 50% белков изменения генетической природы: (первичная структура) приводят к гибели клетки, и мутация не реализуется в наследственную болезнь. Такие белки называются мономорфными. Они обеспечивают основные функции клетки, консервативно сохраняя стабильность видовой организации клетки. Так или иначе, но число генных болезней действительно большое. В настоящее время оно исчисляется тысячами (около 4,5 тыс.).
В группу генных болезней входят аутосомные болезни (доминантные и рецессивные) и сцепленные с полом (доминантные и рецессивные).
При доминантных аутосомных заболеваниях патологический ген находится в аутосоме и проявляет себя даже в гетерозиготном состоянии.
Особенности передачи доминантных аутосомных заболеваний следующие:
1. Лица мужского и женского пола поражаются в равной степени.
2. Передача патологического признака возможна от любого из родителей.
3. Частота индивидуального поражения среди потомков больного, как правило, составляет 50%.
4. Встречаются в каждом поколении (при условии 100% пенетрантности).
Аутосомно-доминантно наследуются: короткопалость, брахидактилия, многопалость, множественного полипоз кишечника, врожденный птоз век, ахондроплазия (рис. 5-4), врожденная куриная слепота (не поддающаяся лечению витамином А), болезнь Марфана (портрет Линкольна, арахнодактилия - паучьи пальцы, подвывих хрусталика и др.), хорея Гентингтона (проявляется в 35-40 лет, имеет 2 основных синдрома: хорея - гиперкинетические подергивания туловища, лица, шаркающая походка, симптом нарушения речи из-за подергивания языка и нёба; деменция - слабоумие) и др. Экспрессивность при хорее Гентингтона может варьировать от нистагма (толчкообразные движения глаз вертикально, в стороны, вращательно) до полной деменции (слабоумия), что свидетельствует о клиническом полиморфизме наследственных заболеваний.
Аутосомные рецессивные генные болезни проявляются только в гомозиготном состоянии, патологический ген находится в аутосоме.
Особенности передачи рецессивных аутосомных заболеваний таковы:
1. Лица мужского и женского пола поражаются в равной степени.
2. Как правило, родители больного фенотипически здоровы, являются гетерозиготами, носителями патологического гена.
3. При этом риск рождения больного ребенка - 25%.
4. Если болен один из родителей, дети обычно здоровы.
5. Нередко родители больного ребенка являются родственниками (выше вероятность быть носителями одного и того же рецессивного гена).
Аутосомно-рецессивно наследуются энзимопатии - наследственные дефекты обмена углеводов (пример - галактоземия), липидов (пример - сфинголипидозы), аминокислот (примеры - фенилкетонурия, альбинизм); витаминов, эритроцитарных ферментов, дефекты биосинтеза гормонов, коллагеновые болезни. Аутосомно-рецессивно наследуется муковисцидоз - заболевание, характеризующееся образованием в железах густого секрета, который закупоривает железистые протоки, в результате формируются кисты. Выделяют легочную и кишечную форму муковисцидоза.
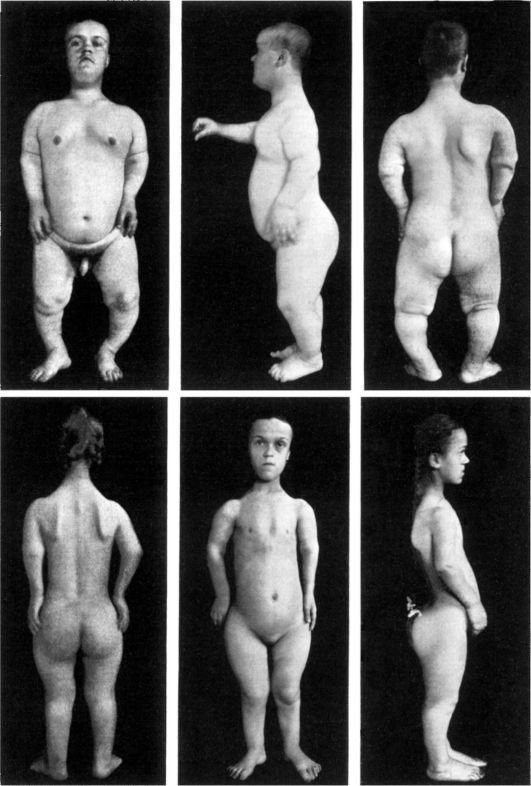 Рис. 5-4. Брат и сестра с ахондроплазией (по W. Falta, 1913)
Рис. 5-4. Брат и сестра с ахондроплазией (по W. Falta, 1913)
Например, в гене муковисцидоза описано свыше 900 вызывающих болезнь мутаций следующих типов: делеции, миссенс, нонсенс, сдвиг рамки считывания, нарушения сплайсинга. Более 400 патологических мутаций известно для гена фенилкетонурии (миссенс, нонсенс, делеции, нарушения сплайсинга). Аутосомно-рецессивно наследуется болезнь Вильсона-Коновалова (гепатоцеребральная дистрофия). При этом заболевании увеличивается накопление меди в печени, головном мозгу, роговице глаз, что приводит к развитию цирроза печени, артритов, катаракт, неврологических нарушений, гемолитической анемии. В крови при этом наблюдается гипокупремия.
Особенности передачи Х-сцепленных доминантных болезней:
1. Поражаются и мужчины, и женщины. Больных женщин в 2 раза больше, чем больных мужчин.
2. Все дочери больного отца будут больными, сыновья здоровы.
3. Если мать гомозиготна по данному признаку, то все потомство будет больным, если гетерозиготна - больными будут 50% детей (сыновей и дочерей).
4. В среднем гетерозиготные женщины болеют менее тяжело, чем гемизиготные мужчины.
Примеры заболеваний, которые наследуются Х-сцепленно доминантно: дефект зубной эмали, аномалия волосяных фолликулов (фолликулярный гиперкератоз, приводит к полной или частичной утрате ресниц, бровей, волос головы, тяжелые формы развиваются только у мужчин) и др. Х-сцепленно доминантно наследуется гипофосфатемический рахит - нарушается реабсорбция фосфатов канальцами почек; у мальчиков наблюдаются гипофосфатемия, карликовость, рахит, у девочек - только гипофосфатемия.
Особенности передачи Х-сцепленных рецессивных болезней:
1. Передача патологического гена происходит от отца дочери, все дочери больного отца - фенотипически здоровые носители.
2. Женщина-носитель передаст патологический ген детям с вероятностью 50%.
3. Больной мужчина может получить патологический ген только от матери.
4. Женщина-носитель может получить патологический ген как от матери, так и от отца.
5. Женщины болеют редко. Рождение больной дочери возможно только в случае брака гемизиготного отца и гетерозиготной матери, происходит гомозиготирование - заболевание протекает
тяжело, часть плодов абортируется, часть новорожденных погибает на первом году жизни.
6. У гомозиготной же больной матери будут больными только сыновья, дочери будут носителями (в случае отсутствия патологического гена у отца).
Примеры Х-сцепленных рецессивных болезней: гемофилия А, гемофилия В; дальтонизм, сцепленный с полом ихтиоз, агаммаглобулинемия - болезнь Бруттона, недостаток глюкозо- 6-фосфатдегидрогеназы (Г-6-ФДГ), синдром Леша-Нихана - редкая аномалия метаболизма пуринов, связанная с недостаточностью фермента гипоксантин-гуанин-фосфорибозилтрансферазы, характеризуется тяжелой гиперурикемией, неврологическими расстройствами, идиотией, неукротимым стремлением к самоповреждениям - откусыванию пальцевых фаланг, кончика языка. Х-сцепленно рецессивно наследуется болезнь Менкеса (болезнь «курчавых волос») - нарушение метаболизма меди, проявляется тяжелыми поражениями ЦНС.
Особенностями передачи Y-сцепленных признаков является то, что признак передается от отца всем мальчикам. На Y-хромосоме локализован ряд генов, детерминирующий развитие семенников, отвечающих за сперматогенез, контролирующий интенсивность роста тела, конечностей и зубов, определяющий оволосение ушной раковины. Естественно, патологические мутации, затрагивающие формирование семенников и сперматогенез, наследоваться не могут, потому что такие индивиды стерильны. В последнее время установлено, что на Y-хромосоме расположен ген SRY, ответственный за дифференцировку пола. При генотипе XY в случае нарушения гена SRY может развиться женский фенотип. Транслокация гена SRY на Х-хромосому может привести к рождению мужчины с кариотипом ХХ. Для полного развития мужского фенотипа достаточно наличия в геноме только гена SRY, а не целой Y-хромосомы.
Патогенез генных болезней связан с первичным эффектом мутантного аллеля. Поэтому принципиальные позиции патогенеза генных болезней можно представить следующим образом: мутантный аллель - патологический первичный продукт (качественно или количественно) - цепь последующих биохимических процессов - клетки - органы - организм.
Это и есть главная общая закономерность развития генных болезней при всем их многообразии. В зависимости от того, какой
продукт контролируется конкретным геном и каков характер нарушения его при мутации, соответствующим образом развертывается патогенез болезни на молекулярном уровне.
Первичные эффекты любых (ядерных и митохондриальных) мутантных аллелей могут проявляться в четырех вариантах: 1) количественно избыточном синтезе полипептидной цепи (белка); 2) синтезе аномальной по первичной структуре полипептидной цепи (белка); 3) отсутствии синтеза полипептидной цепи (белка); 4) количественно недостаточном синтезе полипептидной цепи (белка). На основе первичного эффекта мутантного аллеля (при каждой болезни он всегда один и тот же) уже развертывается весь сложнейший патогенез генной болезни, проявляющийся в разнообразных фенотипических эффектах или клинической картине заболевания.
Если в результате мутации вырабатывается избыточное количество белка, то весь патогенез болезни в целом будет обусловлен именно гиперпродукцией генной активности. Так, при первичном гемохроматозе синтезируется избыточное количество глобина, что приводит к перенагруженности эритроцитов гемоглобином и соответственно железом. Увеличивается свертываемость крови, развивается гемосидероз паренхиматозных органов.
При другом варианте патологического эффекта мутантного гена вырабатывается аномальный белок. За этим следуют функциональные нарушения в системе, работу которой в физиологических условиях обеспечивает белок с нормальной структурой. Нарушения эти первоначально развертываются на молекулярном уровне. Примером такого варианта патогенеза болезни может быть серповидно-клеточная анемия. Замена гидрофильной глутаминовой кислоты на гидрофобный валин в структуре глобина изменяет функциональные свойства гемоглобина (пониженная растворимость, повышенная полимеризация). Он не может выполнять кислородакцепторную функцию и кристаллизуется при недостатке кислорода. В результате эритроциты приобретают серповидную форму (отсюда и название болезни), склеиваются и тромбируют капилляры и т.п.
Третий вариант патологического эффекта мутантного аллеля - отсутствие выработки первичного продукта. Это выражается в накоплении токсических продуктов-предшественников. Например, при фенилкетонурии в крови накапливается фенилаланин, поскольку из-за отсутствия фенилаланингидроксилазы печени он не
превращается в тирозин. Могут использоваться другие (обходные) пути обмена, часто также с патологическим исходом. В результате отсутствия первичного продукта гена может задерживаться какойлибо важный процесс, постоянно осуществляющийся в организме. Так, мутации генов, детерминирующих ферменты репарации ДНК, делают невозможным восстановление постоянно возникающих нарушений в структуре ДНК, что приводит к развитию злокачественных новообразований (пигментная ксеродерма, атаксиятелеангиэктазия).
Четвертый вариант первичного патологического эффекта мутантного аллеля - это недостаточность выработки нормального первичного продукта. Примерами являются гипокаталаземия (низкий уровень каталазы в крови, сопровождающийся рецидивирующими инфекциями, изъязвлением десен и слизистой оболочки полости рта) и β-талассемия (дефицит синтеза β-цепей глобина, обусловливающий нестабильность молекулы гемоглобина, укорочение жизни эритроцитов и развитие гемолитической анемии).
Выше были описаны общие закономерности патогенеза генных болезней на молекулярном уровне на примерах нарушения обмена веществ. Тот же самый принцип патогенеза (мутантный аллель - патологический первичный продукт) действует и для генов морфогенетического контроля, мутации в которых приводят к врожденным порокам развития (полидактилия, синдромы Холт- Орама, Нунена, Лоуренса-Муна, Меккеля, Робертса). Начальное звено врожденного порока развития связано с нарушением дифференцировки клеток. Запрограммированная в геноме дифференцировка клеток, а затем и органогенез осуществляются путем смены активации и выключения определенных генов в строго ограниченных временных (по отношению к онтогенезу) промежутках. Если первичный продукт морфогенетического гена аномальный, то необходимая для дальнейшего правильного развития органа дифференцировка клеток не последует. Естественно, что морфогенетических генов много, действуют они в разные периоды онтогенеза. Соответственно, мутации в них будут приводить к специфическим врожденным порокам развития.
Патогенез генных болезней не заканчивается на молекулярном уровне даже в его первичных звеньях. Для многих болезней основным звеном патогенеза является клеточный уровень. При этом речь идет не только о биологической аксиоме: во всех генетических процессах клетка является дискретной, самостоятельно регулируе-
мой единицей, и в ней осуществляются все процессы реализации генетической информации (транскрипция, трансляция, синтез белка). Клеточный уровень патогенеза генных болезней означает, что в определенных типах клеток разыгрываются основные патологические процессы, характерные для конкретной нозологической формы. Клетка как бы «не выпускает из себя патологические явления, а принимает удар от первичного патологического эффекта гена на себя». Точкой приложения являются отдельные структуры клетки, различные при разных болезнях (лизосомы, пероксисомы, мембраны).
Патогенез на клеточном уровне развертывается при болезнях накопления в связи с нарушением ферментативной активности в лизосомах. Так, накопление в клетках гликозоаминогликанов (мукополисахаридов), а в последующем и в основном межклеточном веществе приводит к развитию тяжелой группы заболеваний - мукополисахаридозов. Причиной избыточного содержания полимеров - гликозоамингликанов - является отсутствие их деградации в лизосомах (рис. 5-5, см. цв. вклейку). Нарушение распада гликозоамингликанов связано с дефектами в группе специфических ферментов, осуществляющих весь цикл деградации.
Другим примером болезней накопления могут быть гликогенозы. В клетках печени, почек, мышц и др. накапливаются полимеры гликогена, которые не подвергаются деградации даже тогда, когда организму необходима глюкоза в крови. Например, при гликогенозе I типа наследственный дефект фермента глюкозо-6- фосфатазы приводит к накоплению гликогена в печени и почках, глюкоза в кровь не поступает.
Другие внутриклеточные структуры - пероксисомы - также могут являться точкой приложения первичного действия мутантного гена. В этих случаях развиваются так называемые пероксисомные болезни. К этой группе относятся синдром Цельвегера, ризомелическая точечная остеохондродисплазия, болезнь Рефсума, акаталазия и др.
Мембраны клеток также могут быть ключевыми элементами патогенеза генных болезней. Так, нарушение синтеза рецепторов андрогенов приводит при хромосомном наборе ХУ к развитию женского фенотипа, наличию семенников в брюшной полости и повышенному уровню андрогенов (синдром тестикулярной феминизации). Клиника витамин D-резистентного рахита (аутосомнодоминантное заболевание) обусловлена дефектом рецепторов 1,25-
дигидроксихолекальциферолла. Патологические мутации (табл. 5-2) в гене рецептора липопротеинов низкой плотности (ЛПНП) вызывают дефекты клеточного рецептора. Эффект любой мутации - нарушение гомеостаза липидного обмена.
Таблица 5-2. Эффекты мутаций в гене рецептора ЛПНП на генный продукт
| Класс мутаций | Последствия |
| Нулевой | Нет белка-рецептора |
| Дефект транспорта | Отсутствует или уменьшено число рецепторов на клеточной поверхности |
| Дефект связывания | Нормальное число рецепторов, но отсутствует или уменьшено связывание ЛПНП |
| Дефект интернализации | Нормальное число рецепторов и связывание ЛПНП; отсутствие или снижение эндоцитоза ЛПНП |
| Дефект возвращения | Нормальное число рецепторов, связывание ЛПНП и эндоцитоз; отсутствует или уменьшено освобождение ЛПНП и возвращение рецептора на клеточную поверхность |
При муковисцидозе (синоним - кистозный фиброз) нарушается регуляция транспорта хлоридов через мембраны эпителиальных клеток, которая в норме осуществляется белком - продуктом гена, названным «кистофиброзным трансмембранным регулятором» (сокращенно CFTR). Одни мутации в гене CFTR ведут к снижению синтеза белка CFTR из-за незавершенности процессинга РНК, другие - к качественным изменениям мембранных хлоридных каналов. Одна первичная биохимическая аномалия (нарушение транспорта хлоридов) ведет к мультиорганному патологическому процессу (прогрессирующее поражение дыхательных путей, хронические синуситы, недостаточность экзогенной секреторной функции поджелудочной железы, стерильность у мужчин).
Клеточный уровень патогенеза генных болезней может проявляться не только в конкретных органеллах, но и в виде нарушения скоординированности функций интерфазного периода клетки и размножения. Так, мутации, затрагивающие области онкогенов,
ведут к снятию контроля размножения клеток (репрессия антионкогенов) и соответственно к злокачественному росту (наследственные раки толстого кишечника, ретинобластома).
Клетка может быть главным звеном при реализации молекулярного уровня патогенеза. Так, прекращение синтеза мышечного белка дистрофина при мутациях в этом гене приводит к постепенной деградации мышечных клеток. Это и является основным звеном патогенеза тяжелой наследственной болезни - миопатии Дюшенна.
Органный уровень патогенеза наследственных болезней, безусловно, является производным от молекулярного и клеточного. При различных болезнях мишенью патологического процесса служат разные органы - иногда в результате первичных процессов, иногда - вторичных. Так, отложение меди в печени и в экстрапирамидной системе мозга при гепато-лентикулярной дегенерации (болезнь Вильсона) является первичным процессом, а гемосидероз паренхиматозных органов при первичном гемохроматозе или талассемии развивается вторично вследствие усиленного распада эритроцитов. При алкаптонурии отложения гомогентизиновой кислоты в хрящах суставных поверхностей и клапанах сердца являются вторичным процессом, обусловленным высокой концентрацией гомогентизиновой кислоты в крови (она не превращается в малеилацетоуксусную кислоту в результате мутационно обусловленного отсутствия оксидазы гомогентизиновой кислоты). Это медленно ведет (примерно к 40 годам) к порокам сердца и тугоподвижности суставов.
В организме в целом взаимосвязь патогенетических процессов проявляется сочетанно на молекулярном, клеточном и органном уровнях. Патологический процесс, индуцированный первичным эффектом мутантного аллеля, приобретает целостность с закономерными межиндивидуальными вариациями. Тяжесть и скорость развития болезни при прочих равных условиях (пол ребенка, одинаковый характер мутации) зависят от генотипа организма (генымодификаторы) и условий среды. Патогенез любой наследственной болезни у разных индивидов, хотя и сходен по первичным механизмам и этапам, формируется строго индивидуально.

