




Изотермы Ван-дер-Ваальса
Проанализируем изотермы уравнения Ван–дер–Ваальса – зависимости Р от V для реального газа при постоянной температуре. Умножив уравнение Ван-дер-Ваальса на V 2 и раскрыв скобки, получаем
PV 3 – (RT + bP) vV 2 + av2V - abv3 = 0.
Поскольку данное уравнение имеет третью степень относительно V, а коэффициенты при V действительны, то оно имеет либо один, либо три вещественных корня, т.е. изобара Р = const пересекает кривую Р = Р(V) в одной или трех точках, как это изображено на рисунке 7.4. Причем с повышением температуры мы перейдем от немонотонной зависимости Р = Р(V) к монотонной однозначной функции. Изотерма при Ткр, которая разделяет немонотонные T < Tкр и монотонные T > Ткр изотермы, соответствует изотерме при критической температуре. При температуре выше критической зависимость Р = Р(V) является однозначной монотонной функцией объема. Это означает, что при T > Ткр вещество находится только в одном, газообразном состоянии, как это имело место у идеального газа. При температуре газа ниже критической такая однозначность исчезает, а это означает возможность перехода вещества из газообразного в жидкое и наоборот. На участке АСВ изотермы Т1 давление растет с увеличением объема (dP/dV) > 0. Данное состояние неустойчиво, поскольку здесь должны усиливаться малейшие флуктуации плотности. Поэтому область ВСА не может устойчиво существовать. В областях DLB и AGE давление падает с увеличением объема (dP/dV)Т < 0 – это необходимое, но не достаточное условие устойчивого равновесия. Эксперимент показывает, что система переходит из области устойчивых состояний GE (газ) в область устойчивых состояний LD (жидкость) через двухфазное состояние (газ – жидкость) GL вдоль горизонтальной изотермы GCL.
При квазистатическом сжатии, начиная с точки G, система распадается на 2 фазы – жидкость и газ, причем плотности жидкости и газа остаются при сжатии неизменными и равными их значениям в точках L и G соответственно. При сжатии количество вещества в газообразной фазе непрерывно уменьшается, а в жидкой фазе – увеличивается, пока не будет достигнута точка L, в которой все вещество перейдет в жидкое состояние.
 Рис. 7.4
Наличие критической точки на изотерме Ван–дер–Ваальса означает, что для каждой жидкости существует такая температура, выше которой вещество может существовать только в газообразном состоянии. К этому заключению пришел и Д.И. Менделеев в 1861 г. Он заметил, что при определенной температуре прекращалось поднятие жидкости в капиллярах, т.е. поверхностное натяжение обращалось в нуль. При той же температуре обращалась в нуль скрытая теплота парообразования. Такую температуру Менделеев назвал температурой абсолютного кипения. Выше этой температуры, согласно Менделееву, газ не может быть сконденсирован в жидкость никаким увеличением давления.
Критическую точку K мы определили как точку перегиба критической изотермы, в которой касательная к изотерме горизонтальна (рис. 7.5). Ее можно определить также как точку, в которую в пределе переходят горизонтальные участки изотерм при повышении температуры до критической. На этом основан способ определения критических параметров Pk, Vk, Тk, принадлежащий Эндрюсу. Строится система изотерм при различных температурах. Предельная изотерма, у которой горизонтальный участок LG (рис. 7.4) переходит в точку, будет критической изотермой, а указанная точка – критической точкой (рис. 7.5). Рис. 7.4
Наличие критической точки на изотерме Ван–дер–Ваальса означает, что для каждой жидкости существует такая температура, выше которой вещество может существовать только в газообразном состоянии. К этому заключению пришел и Д.И. Менделеев в 1861 г. Он заметил, что при определенной температуре прекращалось поднятие жидкости в капиллярах, т.е. поверхностное натяжение обращалось в нуль. При той же температуре обращалась в нуль скрытая теплота парообразования. Такую температуру Менделеев назвал температурой абсолютного кипения. Выше этой температуры, согласно Менделееву, газ не может быть сконденсирован в жидкость никаким увеличением давления.
Критическую точку K мы определили как точку перегиба критической изотермы, в которой касательная к изотерме горизонтальна (рис. 7.5). Ее можно определить также как точку, в которую в пределе переходят горизонтальные участки изотерм при повышении температуры до критической. На этом основан способ определения критических параметров Pk, Vk, Тk, принадлежащий Эндрюсу. Строится система изотерм при различных температурах. Предельная изотерма, у которой горизонтальный участок LG (рис. 7.4) переходит в точку, будет критической изотермой, а указанная точка – критической точкой (рис. 7.5).
 Рис. 7.5
Недостаток способа Эндрюса заключается в его громоздкости. Рис. 7.5
Недостаток способа Эндрюса заключается в его громоздкости.
|
Критическая температура, 1) температура вещества в его критическом состоянии. Для индивидуальных веществ Критическая температура определяется как температура, при которой исчезают различия в физических свойствах между жидкостью и паром, находящимися в равновесии. При Критическая температура плотности насыщенного пара и жидкости становятся одинаковыми, граница между ними исчезает, и теплота парообразования обращается в нуль. К. т. - одна из неизменяющихся характеристик (констант) вещества. Значения Критическая температура Tk некоторых веществ приведены в ст. Критическая точка.
В двойных системах (например, пропан - изопентан) равновесие жидкость - пар имеет не одну Критическая температура, а пространственную критическую кривую, крайними точками которой являются Критическая температура чистых компонентов.
2) температура, при которой в жидких смесях с ограниченно растворимыми компонентами наступает их взаимная неограниченная растворимость; её называют Критическая температура растворимости.
3) температура перехода ряда проводников в сверхпроводящее состояние (см. Сверхпроводимость). Измерена у большого числа металлов, сплавов и химических соединений. В чистых металлах наинизшая Критическая температура наблюдается у Ti (0,37 К), самая высокая - у Тс (11,2 К). Очень высокое значение Критическая температура найдено у сплава Nb, Al и Ge (Tk»21 К).
Внутренняя энергия газа Ван-дер-Ваальса
Энергия одного моля газа Ван–дер–Ваальса слагается из внутренней энергии молекул, составляющих газ: кинетической энергии теплового движения центра масс молекул 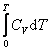 , равной, и потенциальной энергии взаимного притяжения молекул.
Потенциальная энергия притяжения молекул равна работе, необходимой для разведения молекул на бесконечное расстояние друг от друга. В этом конечном состоянии молекулы не взаимодействуют друг с другом, а потенциальную энергию можно считать равной нулю. Дополнительное давление газа Ван-дер-Ваальса за счет взаимного притяжения молекул равно a/Vm2 и, следовательно, потенциальная энергия взаимодействия равна: , равной, и потенциальной энергии взаимного притяжения молекул.
Потенциальная энергия притяжения молекул равна работе, необходимой для разведения молекул на бесконечное расстояние друг от друга. В этом конечном состоянии молекулы не взаимодействуют друг с другом, а потенциальную энергию можно считать равной нулю. Дополнительное давление газа Ван-дер-Ваальса за счет взаимного притяжения молекул равно a/Vm2 и, следовательно, потенциальная энергия взаимодействия равна:
 .
Знак «минус» указывает на то, что между молекулами действуют силы притяжения; Vm – молярный объем, Vm = V/µ, v = m/m.
Полная энергия одного моля газа Ван-дер-Ваальса определяется соотношением .
Знак «минус» указывает на то, что между молекулами действуют силы притяжения; Vm – молярный объем, Vm = V/µ, v = m/m.
Полная энергия одного моля газа Ван-дер-Ваальса определяется соотношением
 .
Если СV не зависит от температуры, то имеем для одного моля
Um = CV Т– a/Vm.
Причиной недостаточной точности уравнения Ван–дер–Ваальс считал ассоциацию молекул в газовой фазе, которую не удается описать, учитывая зависимость параметров a и b от объема и температуры, без использования дополнительных постоянных. После 1873 г. сам Ван–дер–Ваальс предложил еще шесть вариантов своего уравнения, последнее из которых относится к 1911 г. и содержит пять эмпирических постоянных. Две модификации уравнения предложил Клаузиус, и обе они связаны с усложнением вида постоянной b. Больцман получил три уравнения этого типа, изменяя выражения для постоянной a. Всего известно более сотни подобных уравнений, отличающихся числом эмпирических постоянных, степенью точности и областью применимости. Выяснилось, что ни одно из уравнений состояния, содержащих менее 5 индивидуальных постоянных, не оказалось достаточно точным для описания реальных газов в широком диапазоне Р, V, T, и все эти уравнения оказались непригодными в области конденсации газов. Из простых уравнений с двумя индивидуальными параметрами неплохие результаты дают уравнения Дитеричи и Бертло.
Принципиальное значение уравнения Ван–дер–Ваальса определяется следующими обстоятельствами:
уравнение было получено из модельных представлений о свойствах реальных газов и жидкостей, а не явилось результатом эмпирического подбора функции f(p,V,T), описывающей свойства реальных газов;
уравнение долго рассматривалось как некоторый общий вид уравнения состояния реальных газов, на основе которого было построено много других уравнений состояния;
с помощью уравнения Ван–дер–Ваальса впервые удалось описать явление перехода газа в жидкость и проанализировать критические явления. В этом отношении уравнение Ван–дер–Ваальса имеет преимущество даже перед более точными уравнениями в вириальной форме. .
Если СV не зависит от температуры, то имеем для одного моля
Um = CV Т– a/Vm.
Причиной недостаточной точности уравнения Ван–дер–Ваальс считал ассоциацию молекул в газовой фазе, которую не удается описать, учитывая зависимость параметров a и b от объема и температуры, без использования дополнительных постоянных. После 1873 г. сам Ван–дер–Ваальс предложил еще шесть вариантов своего уравнения, последнее из которых относится к 1911 г. и содержит пять эмпирических постоянных. Две модификации уравнения предложил Клаузиус, и обе они связаны с усложнением вида постоянной b. Больцман получил три уравнения этого типа, изменяя выражения для постоянной a. Всего известно более сотни подобных уравнений, отличающихся числом эмпирических постоянных, степенью точности и областью применимости. Выяснилось, что ни одно из уравнений состояния, содержащих менее 5 индивидуальных постоянных, не оказалось достаточно точным для описания реальных газов в широком диапазоне Р, V, T, и все эти уравнения оказались непригодными в области конденсации газов. Из простых уравнений с двумя индивидуальными параметрами неплохие результаты дают уравнения Дитеричи и Бертло.
Принципиальное значение уравнения Ван–дер–Ваальса определяется следующими обстоятельствами:
уравнение было получено из модельных представлений о свойствах реальных газов и жидкостей, а не явилось результатом эмпирического подбора функции f(p,V,T), описывающей свойства реальных газов;
уравнение долго рассматривалось как некоторый общий вид уравнения состояния реальных газов, на основе которого было построено много других уравнений состояния;
с помощью уравнения Ван–дер–Ваальса впервые удалось описать явление перехода газа в жидкость и проанализировать критические явления. В этом отношении уравнение Ван–дер–Ваальса имеет преимущество даже перед более точными уравнениями в вириальной форме.
|

