




Очень важным вопросом, непосредственно зависящим от наличия двойного электрического слоя, является устойчивость дисперсных систем. Проблема устойчивости, или как её часто эмоционально называют “проблема жизни и смерти дисперсных систем”, это одна из наиболее интересных и востребованных современной наукой проблем коллоидной химии. Можно привести массу примеров, когда есть необходимость сохранить устойчивость дисперсной системы, и не меньшее число примеров, когда, напротив, требуется эту устойчивость нарушить. Обеспечение устойчивости свободно-дисперсных систем необходимо при получении из них различных покрытий, лекарственных препаратов, аэрозольных средств, связующих материалов и т.п. Если мы имеем дело с полировальной композицией, содержащей частицы абразивного материала нанометрового размера, то вполне понятно, что слипание этих частиц в более крупные агрегаты во время проведения технологического процесса, сделает дальнейшее использование композиции неприемлемым для выполнения поставленной задачи. С другой стороны, ликвидация устойчивости дисперсных систем требуется для получения осадков при разделении фаз, при очистке промышленных выбросов, для обеспечения процесса структурообразования при получении строительных материалов и т.д. Если речь идет об организации замкнутого цикла малоотходных производств, например о регенерации сточных вод, то для их очистки сначала необходимо провести процесс коагуляции, т.е. слипания частиц дисперсной фазы в более крупные агрегаты, которые затем можно будет отфильтровать обычными фильтрами.
Устойчивость дисперсной системы характеризуется постоянством во времени двух основных параметров этой системы: 1) исходной степени дисперсности (первоначальных размеров частиц) ― агрегативная устойчивость; 2) равновесного распределения частиц дисперсной фазы по объему дисперсионной среды ― седиментационная устойчивость. Нас сейчас интересует именно агрегативная устойчивость, т.е. способность дисперсной системы сохранять исходный размер частиц.
Когда же мы имеем дело с двумя лиофобными частицами одного знака заряда, окруженными двойными электрическими слоями (ДЭС), с одинаковыми по заряду противоионами в диффузных частях ДЭС, то действием, приводящим к потери устойчивости, будет сжатие диффузной части ДЭС,которое достигается введением в эту систему сильных электролитов.
Тот факт, что все сильные электролиты вызывают коагуляцию, был известен уже давно. Более 100 лет тому назад было сформулировано эмпирическое правило Шульце – Гарди:
“Коагулирующий ион имеет заряд, противоположный по знаку заряду твердой фазы, и порог коагуляции Ск тем меньше, чем выше заряд этого иона”.
(порог коагуляции Ск ─ это минимальная концентрация электролита, вызывающая коагуляцию).
Согласно правилу Шульце – Гарди соотношение порогов коагуляции для ионов с различной величиной заряда выглядит приблизительно, как СI: СII: СIII ≈ 500: 8: 1
В зависимости от химической природы иона пороги коагуляции также различаются, причем они в принципе повторяют лиотропный ряд, известный для адсорбционной способности этих ионов, т.е. Ск возрастает в ряду:
Al+3 < Ba+2 < Mg+2 < Rb+1 < K+1 < Na+1.
Чем меньше, по мере увеличения концентрации электролита в дисперсной системе, будут становиться абсолютные величины (│ ψ1 │и│ ζ │) - потенциалов, тем сильнее происходит сжатие ДЭС, иследовательно, тем ближе могут подойти друг к другу две частицы, в результате чего между ними возникнет непосредственный контакт и образуется единый агрегат из пары частиц, т.е. пройдет коагуляция. Для осуществления такого процесса абсолютная величина ζ– потенциала не обязательно должна быть равна нулю, (как это предполагалв 1900 г. Гарди). Вполне достаточно, чтобы она была невелика, меньше некоторого критического значения, порядка 20 – 25 мВ.
ζ (дзета) – потенциал – это часть падения потенциала в двойном электрическом слое на границе раздела твердой фазы с раствором электролита, которая относится к движущейся части ДЭС, и величина которой может быть определена с помощью электрокинетических явлений.
Рсаамотрим график зависимости ζ – потенциала от концентрации различных индифферентных электролитов, проведя на нем две пунктирные линии, параллельные оси концентраций, обозначающие критические значения ζ– потенциала, в области как отрицательных, так и положительных величин ζ (рис. XI.2).

Кривая 1 соответствует зависимости ζ от lgС для KNO3. При всех концентрациях KNO3 меньших, чем СkI, система остается устойчивой, т.к. при этих концентрациях абсолютная величина│ ζ │ выше критического значения, а при достижении концентрации СkI начинается коагуляция, которая сохраняется и при дальнейшем росте концентрации KNO3. Аналогично ведет себя система при добавлении Ba(NO3 )2 ,кривая 2: устойчивость при концентрациях электролита меньше величины CkII и окончательная коагуляция при больших концентрациях Ba(NO3 )2. Зато совсем иная картина наблюдается в случае коагуляции раствором Al(NO3)3, кривая 3,т.е. когда коагулирующим является многозарядный противоион Al+3. За счет сверхэквивалентного содержания «+» заряда уже в плотном слое, катион Al+3 вызывает перезарядку поверхности частицы, с переменой знака (ψ1 и ζ) – потенциалов, (вспомните рис X.9 из главы X). Поэтому при очень малых концентрациях Al(NO3)3, пока ζ – потенциал ещё не достиг критического значения наблюдается первая зона устойчивости, затем в интервале концентраций, соответствующих значениям ζ между ─ ζ кр и + ζ кр , появляется первая зона коагуляции.
Однако, при дальнейшем увеличении концентрации электролита, величина + ζ снова становится больше, чем + ζкр, что приводит к появлению второй зоны устойчивости. Ещё большее увеличение концентрации Al(NO3)3 приводит к новому понижению значений ζ – потенциала до величин более низких, чем + ζ кр,и к появлению второй (и теперь уже окончательной) зоны коагуляции.
Исследование процесса коагуляции можно проводить различными методами.
Наиболее информативным прямым методом исследования является метод счёта частиц в дисперсной системе (золе) с помощью поточного ультрамикроскоп
|
хорошо знакомый нам золь AgI c отрицательным зарядом частиц и меняя в нём концентрацию коагулирующего электролита Al(NO3)3, мы сможем получить график зависимости мутности этого золя τ от концентрации электролита lgc, представленный на рис. XI.9
При сравнении этой зависимости с рис. XI.2 [нижняя кривая для Al(NO3)3 ]отчетливо видны те же две зоны устойчивости и две зоны коагуляции.
В данной работе изучают коагуляцию золя AgI раствором Al(NO3)3 в области концентраций от 1* 10-6 до 3*10-3 моль/л. Золь получают непосредственно в кювете, в которой затем производится измерение оптической плотности D5.
В кювету наливают в строго определенном порядке: 1) дистиллированную воду; 2) раствор Al(NO3)3; 3) 10 мл раствора AgNO3; 4) 10 мл раствора KI так, чтобы общий объем растворов в кювете составил 21 мл. Растворы AgNO3 и KI необходимой концентрации находятся на рабочем месте в бюретках, растворы Al(NO3)3 с концентрациями 2.1*10-4; 2.1*10-3; 2.1*10-2 в трех отдельных пробирках пронумерованных, по возрастанию концентрации. Общий объем воды и раствора Al(NO3)3 составляет 1 мл, а соотношение между ними задано в табл. 2

Рис. XI.9 Зависимость мутности τ золя AgI от концентрации коагулирующего электролита Al(NO3)3.
Требуемое количество воды и раствора Al(NO3)3 рекомендуется вливать в кювету по разности начального и конечного объемов раствора в пипетке. После смешения всех четырех растворов включают секундомер и содержимое кюветы тщательно перемешивают, осторожно продувая воздух через пипетку, носик которой подносят к поверхности раствора в кювете. Затем кювета помещается в фотоэлектроколориметр и измерения оптической плотности D5 производят через 5 (пять) минут после смешения растворов, по три раза подряд. После измерения золь выливают в специальную склянку для слива растворов, содержащих серебро, кювета прополаскивается из промывалки дистиллированной водой и переходят к следующему опыту.
Данные каждых трех измерений усредняют (D5ср), после чего рассчитывают значения мутности золя поформуле τ5 = 2,3 * D5ср / h, где h = 5см – толщина слоя среды, через которую проходит луч в кювете. Измерения оптической плотности проводят на синем светофильтре. Работу начинают с проверки установки нуля, наливая в каждую кювету по 21 мл дистиллированной воды из промывалки. Для выполнения работы выдаются две пипетки: одна для воды, вторая – для раствора Al(NO3)3. Перед началом работы обе пипетки тщательно промывают дистиллированной водой.
Полученные данные заносят в таблицу 1, проверяя каждые пять последовательных измерений оптической плотности у преподавателя. На основании полученных результатов строят график зависимости τ5 от – lgC, аналогичныйпредставленному на рис. XI. 9.
Таблица 1.
| № пробир ки | № опыта | Вода, мл | Объем Al(NO3)3 мл | Концентр. Al(NO3)3 моль/л | Сэл-та в золе, моль/л | -lgC | D5 | D5 ср. | τ5 | |
| I | 1.0 | 0.0 | 2.1*10-4 | H2O | 0.032 0.031 0.033 | 0.032 | 0.0147 | |||
| 0.9 | 0.1 | 1*10-6 | 0.277 0.279 0.278 | 0.278 | 0.1279 | |||||
| 0.8 | 0.2 | 2*10-6 | 5.7 | 0.360 0.361 0.362 | 0.361 | 0.1661 | ||||
| 0.7 | 0.3 | 3*10-6 | 5.5 | 0.170 0.172 0.171 | 0.171 | 0.0787 | ||||
| 0.5 | 0.5 | 5*10-6 | 5.3 | 0.106 0.102 0.104 | 0.104 | 0.0478 | ||||
| 0.0 | 1.0 | 1*10-5 | 0.094 0.092 0.090 | 0.092 | 0.0423 | |||||
| II | 0.8 | 0.2 | 2.1*10-3 | 2*10-5 | 4.7 | 0.092 0.090 0.091 | 0.091 | 0.0421 | ||
| 0.5 | 0.5 | 5*10-5 | 4.3 | 0.165 0.164 0.166 | 0.165 | 0.0759 | ||||
| 0.0 | 1.0 | 1*10-4 | 0.307 0.308 0.309 | 0.308 | 0.1421 | |||||
| 0.8 | 0.2 | 2*10-4 | 3.7 | 0.300 0.305 0.310 | 0.307 | 0.1412 | ||||
| III | 0.7 | 0.3 | 2*10-2 | 3*10-4 | 3.5 | 0.286 0.287 0.288 | 0.287 | 0.1320 | ||
6. Исследование процесса набухания твёрдых полимеров.
Суть работы заключается в определении зависимости степени набухания желатины от времени набухания. Образцы сухой желатины набухают в воде и растворе одного из электролитов (HCl, CH3COOH, KCL, KNO3 и т.п.). Работа выполняется с помощью специальных торзионных весов с масштабом шкалы от 1 до 1000 мг. Получают в лаборантской два стаканчика, две проволочки и два кусочка сухой желатины. Сначала взвешивают две проволочки, на которые затем подвешивают два кусочка сухой желатины, после чего снова взвешивают проволочки с желатиной и таким образом определяют массу каждого из двух кусочков желатины Р0 (для опыта с водой и для опыта с электролитом).
В один стаканчик объемом 50 – 100 мл наливают воду, в другой раствор электролита, после чего опускают кусочки желатины вместе с проволочками в эти стаканчики. Желательно сделать это с интервалом примерно в 5 минут, чтобы в дальнейшем измерения массы набухшей желатины из двух стаканчиков не накладывались друг на друга. Далее через 15 минут вынимают проволочку с кусочком желатины из первого стаканчика, фильтровальной бумагой осторожно подсушивают капельки раствора на поверхности желатины, не пытаясь отжимать его, после чего взвешивают проволочку вместе с уже набухшей желатиной. Через 5 минут проделывают то же самое с образцом из другого стаканчика. Полученные данные записывают в таблицы 1 и 2. Всего проделывают для каждого стаканчика по восемь измерений с интервалом между каждыми двумя измерениями в 15 минут. Работа рассчитана на два часа. По окончании работы выливают растворы из стаканчиков, проволочки возвращают в лаборантскую, желатину выбрасывают.
Образец оформления протокола по работе:
“ Исследование процесса набухания твёрдых полимеров ”
Выполнил студент …(Ф.И.О)… группы ……. курса…….….факультета
По результатам измерения процесса набухания в воде и в данном электролите составляют две таблицы:
Таблица 1. Набухание желатины в KBr.
| Время t, мин | Вес проволоки Рпр, мг | Вес проволоки и сухой желатины Р1 , мг | Вес сухой желатины Р0, мг | Вес проволоки с набухшей желатиной Р2, мг | Вес набухшей желатины Рн=P2 –Pпр, мг | Степень набухания W=(Рн-P0)/P0 |
| 112.5 | 94.5 | 1.35 | ||||
| 94.5 | 1.57 | |||||
| 94.5 | 1.86 | |||||
| 94.5 | 318.5 | 300.5 | 2.18 | |||
| 94.5 | 2.53 | |||||
| 94.5 | 2.80 | |||||
| 94.5 | 3.04 | |||||
| 94.5 | 3.31 |
Таблица 2. Набухание желатины в воде
| Время t, мин | Вес проволоки Рпр, мг | Вес проволоки и сухой желатины Р1 , мг | Вес сухой желатины Р0, мг | Вес проволоки с набухшей желатиной Р2, мг | Вес набухшей желатины Рн=P2-Pпр мг | Степень набухания W=(Рн-P0)/P0 |
| 18.5 | 58.5 | 106.5 | 0.82 | |||
| 58.5 | 132.5 | 1.26 | ||||
| 58.5 | 140.5 | 1.40 | ||||
| 58.5 | 152.5 | 1.61 | ||||
| 58.5 | 188.5 | 1.91 | ||||
| 58.5 | 175.5 | 2.00 | ||||
| 58.5 | 184.5 | 2.15 | ||||
| 58.5 | 195.5 | 2.34 |
По полученным данным строят два графика в координатах: степень набухания W от времени набухания t [ мин.] (мы в качестве примера приводим только один график зависимости W от t для набухания желатины в воде (рис.XI.5).
После того как данные из таблицы 2 нанесены на график (рис.XI.5,), проводят усредненную плавную кривую из начала координат.
Процесс набухания протекает как реакция первого порядка:
dW/dt = A(W∞ - W), где:
А – постоянная, зависящая от природы полимера; W – степень набухания;
W∞ - предельная степень набухания; dW/dt – скорость набухания.
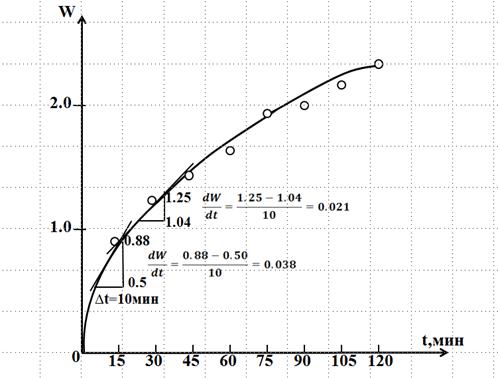
Рис.XIV.5
Можно представить это уравнение в виде W = W∞ − (1/А) * dW/dt (это уравнение прямой, не проходящей через начало координат), с помощью которого можно найти графически предельную степень набухания W∞ и константу А. Для этого необходимо построить график зависимости степени набухания W от скорости набухания dW/dt. Для построения такого графика на кривой (рис. XIV.5) выбирают точки, соответствующие времени набухания 15, 30, 45, 60, 75, 90, 105 мин., и в каждой из этих точек проводят касательную к кривой W = f(t). Используя эту касательную, как гипотенузу, строят треугольники, в каждом из которых катет, соответствующий оси времени, составляет 10 мин. и вычисляют ряд значений ΔW и dW/dt, как это показано на рис .XIV.5.
Затем заполняют таблицу 3 и по её данным строят график зависимости степени набухания W от скорости набухания dW/dt (рис.XIV.6).
Таблица 3.
| t, мин. | |||||||
| W | 0,70 | 1,16 | 1,47 | 1,71 | 1,91 | 2,07 | 2,21 |
| ∆W | 0,88 - 0,50 | 1,26 - 1,04 | 1,54 - 1,35 | 1,77 - 1,64 | 1,96 - 1,85 | 2,13 - 2,03 | 2,24 - 2,17 |
| dW/dt | 0,038 | 0,022 | 0,019 | 0,013 | 0,011 | 0,010 | 0,007 |
По этим точкам (на рис. XIV.6 – звёздочки) проводят усредненную прямую, причем экстраполяция этой прямой на ось ординат (dW/dt = 0), даст значение W∞; а на ось абсцисс (W = 0), даст величину A*W∞ . После этого вычисляют значение А..
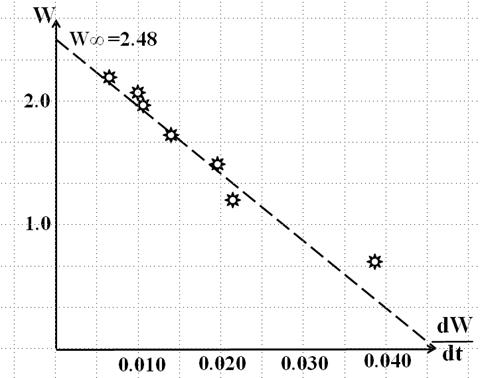
Рис.XIV.6
В нашем случае: W∞ = 2,48; А = 0, 0185.

