




1. В анамнезе следует обратить внимание на состояние здоровья родителей, братьев, сестер, на возраст; отмечалась ли склонность к кровотечениям у родственников (у кого именно, выраженность геморрагического синдрома, число тромбоцитов и т.д.); отмечалась ли у ребенка после рождения геморрагическая сыпь, кровотечения из пупочной ранки, из носа, десен, слизистой ротовой полости и т.д.;
- на перенесенные инфекционные заболевания (особое внимание на эпизоды герпетической инфекции, вирусный гепатит, частые острые респираторные заболевания, ангины, краснуху, ветряную оспу и т.д.);
- на аллергические заболевания (атонический дерматит, крапивница, бронхиальная астма и др.), реакции при проведении профилактических вакцинаций, применении антибиотиков, других лекарственных средств, гемотрансфузиях, введении иммуноглобулина и др.;
- что непосредственно предшествовало началу геморрагического диатеза (инфекционные заболевания, профилактические прививки, нервные потрясения, травмы, погрешности в питании и т.п.);
- на начало и течение пурпуры (когда началось заболевание), выраженность геморрагического синдрома (экхимозы, петехии, кровотечения из носа, ротовой полости, кишечные и др.);
- предшествующее лечение (особенно детализировать гормонотерапию), когда проводилась, дозы максимальные суточные и курсовые, длительность курсов, по особым показаниям - угроза кровоизлияния в мозг или очень тяжелая анемия - гемотрансфузии, переливания тромбоцитарной массы, плазмы и пр. и его результаты; - на причину настоящей госпитализации.
2. В день поступления подробно описать характер геморрагических высыпаний на коже и слизистых оболочках (количество, локализация, симметричность расположения, размеры, цвет и т.п.). Цвет кожи (бледность, желтушность), пигментации от старых кровоизлияний. Данные пальпации селезенки и ее перкуторные размеры. Цвет мочи, кала. Масса и рост ребенка, оценить физическое развитие.
3. В дневниках истории болезни ежедневно описывать состояние кожи и слизистых ооболочек, отмечать новые геморрагии с подробной их характеристикой, эволюцию геморрагических элементов, результаты пальпации печени и селезенки. Артериальное давление измерять не реже одного раза в 3 дня (детям, получающим стероидные гормоны, измеряется ежедневно).
4. В первые дни после поступления ребенок должен быть проконсультирован стоматологом (провести санацию зубов), отоларингологом (хронический тонзиллит и др.).
ГЕМОФИЛИЯ
Среди наследственных коагулопатий, протекающих с нарушением гемостаза, около 96% приходится на два заболевания - гемофилию А и гемофилию В. Гемофилия А - классическая гемофилия, обусловлена дефицитом фактора VIII (антигемофильный глобулин), и гемофилия В (болезнь Кристмаса), связана с дефицитом фактора IX. Соотношение гемофилии А и гемофилии В составляет в среднем 4:1.
Причиной гемофилии могут служить количественные и качественные изменения факторов свертывания крви. Наследственность гемофилии удается установить у 70-90% больных. Кроме наследственной, различают спонтанную форму, которая является следствием возникших мутаций у 10-30% больных гемофилией.
Распространенность гемофилии составляет в большинстве стран 13-14 на 10 000 жителей мужского пола (Якунина Л.Н., 2004).
Ген, регулярующий синтез факторов IХ и VIII, локализуются в Х-хромосомах половых клеток. У женщин, носителей этого заболевания, вторая Х-хромосома нормальная; они как правило, не страдают кровоточивостью, но активность фактора VIII у них снижена в среднем в 2 раза по сравнению с нормальными величинами, и у них может быть кровоточивость во время родов, при операциях, травмах.
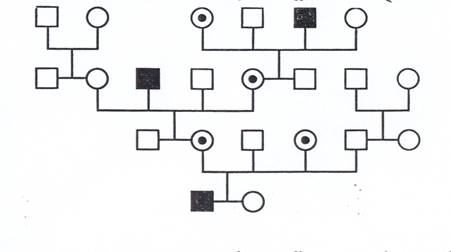 |
По правилам наследования гена, сцепленного с Х-хромосомой, все дочери отца, больного гемофилией, - носители заболевания, все сыновья здоровы. У сыновей, матери которых являются носителями заболевания, вероятность родиться больными составляет 50% (рис. 6).
Рис. 6. Наследование гемофилии. Кружками обозначены женщины;
кружками с точкой - женщины-кондуктор; квадратами - мужчины;
темными квадратами - больные гемофилией.
Женская гемофилия - случаи единичны.
Заключение о возможном носительстве может быть сделано при стабильном снижении концентрации фактора VIII или IX ниже 45%.
Разнообразие форм патологии фактора VIII отражает сложность его структуры. Структура фактора VIII и функциональная характеристика его субъединиц дана в табл. 7.
В норме фактор VIII циркулирует в крови в форме крупномолекулярного полимера. Этот полимер состоит из ряда субъединиц, в состав которых входят гликопротеины, обладающие прокоагулянтной активностью (VIII:К), способностью осуществлять адгезию тромбоцитов, их агглютинацию под влиянием ристомицина и контролировать капиллярное кровотечение, т.е. активность фактора Виллебранда (VIII:ФВ, VIII:Ркоф - ристомициновый кофактор), а также антигенные маркеры VIII:К (VIII:КАг) и белок, тесно связанный с ристомицином-кофактором (VIII:КАг), который рядом авторов рассматривается как белок носитель.
Таблица 7
Компоненты фактора VIII
| Наименование | Сокращенное обозначение | Функциональная характеристика |
| Прокоагулянтная часть | VIII:К (VIII:С) | Обладает антигемофилической активностью; снижен при гемофилии А, взаимодействует с фактором IX; слабоантигенный |
| Антигенный маркер VIII:К | VIII:КАг (VIII:САг) | Взаимодействует с иммунными ингибиторами фактора VIII:К |
| Фактор Виллебранда (кофактор ристомицина) | VIII:ФВ VIII: Ркоф | Контролирует время кровотечения, участвует в адгезии тромбоцитов и в их ристомицин - агрегации, влияет на активность VIII:К в мультимерах |
| Антигенный белок, связанный с VIII:Ркоф | VIII:РАг | Основной антиген комплекса, тесно связанный с фактором Виллебранда и вместе с ним продуцируемый в эндотелии. Возможно, является белком носителем, участвующих в организации мультимеров, и кофактором VIII:К |
Гемофилия А - наследственный диатез, обусловленный наследственным дефицитом или молекулярной аномалией прокоагулянтной части фактора VIII.
Клиника. Тяжесть геморрагических проявлений при гемофилии строго коррелирует со степенью дефицита фактора в плазме, уровень которого в отдельных семьях генетически запрограммирован.
Гемофилию практически полезно подразделять на следующие группы:
а) с уровнем фактора VIII (или IX) от 0 до 1% - крайне тяжелая форма;
б) активность фактора от 1 до 2% - тяжелая форма;
в) от 2 до 5% - форма средней тяжести;
г) с уровнем фактора от 5% до 12% - легкая форма, но с возможностью возникновения тяжелых и даже смертельных кровотечений при травмах и хирургических вмешательствах, проводимых без достаточной заместительной терапии (З.С. Баркаган).
В клинической картине гемофилия характеризуется прежде всего гематомным типом кровоточивости с кровоизлияниями в крупные суставы, глубокими подкожными, межмышечными и внутримышечными гематомами и обильными, длительными кровотечениями при травмах.
Важно отметить возрастные особенности кровоточивости при гемофилии. При рождении в тяжелых случаях наблюдаются обширные кефалогематомы, подкожные и внутрикожные кровоизлияния, поздние кровотечения из пуповинного канатика. Прорезывание зубов сопровождается обычно десневыми кровотечениями. Иногда болезнь выявляется при первой внутримышечной прививке, которая может стать причиной развития большой межмышечной гематомы. В первые годы жизни часты кровотечения из слизистой оболочки ротовой полости, связанные с ее травматизацией различными острыми предметами. Причиной кровотечений бывают также прикусы языка и надрывы его уздечки.
В ползунковом периоде типично появление гематом в области черепа, кровоизлияния в мягкие ткани вблизи глаза, а также заглазничные гематомы, которые могут привести к потере зрения. Однако, у многих больных гемофилией при рождении и в первые годы жизни существенных геморрагических проявлений нет, в связи с чем заболевание распознается лишь на 2-3 году жизни.
Острые гемартрозы появляются тем раньше, чем тяжелее гемофилия. При тяжелых формах в 2-3 летнем возрасте, при формах средней степени тяжести - в 4-6 лет. С наибольшей частотой при гемофилии поражаются коленные суставы, за ними следуют локтевые и голеностопные, затем - лучезапястные, плечевые и тазобедренные, сравнительно редко наблюдаются кровоизлияния в мелкие суставы. У каждого больного имеются суставы, которые с особым упорством и частотой поражаются повторными кровоизлияниями. Связано это с морфологической перестройкой и вторичными воспалительными изменениями тканей сустава - набуханием, складчатостью и интенсивной васкуляризацией синовиальной оболочки, которая вследствие этого легко ущемляется и подвергается травматизации измененными костными и хрящевыми частями сустава.
Клинически важно различать следующие разновидности суставных поражений при гемофилии:
- острые гемартрозы: первичные и рецидивирующие;
- хронические геморрагически-деструктивные остеоартрозы;
Острый гемартроз. Кровоизлияние в сустав проявляется внезапной резкой болью в области сустава. Сустав увеличен в объеме, кожа над ним гиперемирована и горяча на ощупь. При больших кровоизлияниях определяется флюктуация. Характерно быстрое (в течение нескольких часов) ослабление боли после первой же достаточной трансфузии антигемофильных препаратов, и почти немедленное - при одновременной эвакуации крови из сустава. Если болевой синдром при таком лечении не ликвидируется, то следует искать дополнительную патологию - внутрисуставной перелом, отрыв мыщелка, ущемление ткани. Обратное развитие гемартроза происходит на 2-3 неделе.
Хронические остеоартрозы разделяются по стадиям на основе клинико-рентгенологических данных. Принята классификация Э.3. Новиковой (1967), в которой выделяются четыре стадии поражения суставов у больных гемофилией.
В I или ранней стадии может быть увеличен объем сустава (с расширением суставной щели) за счет кровоизлияния. В „холодном" периоде функция сустава не нарушена, но рентгенологически может обнаруживаться утолщение и уплотнение суставной капсулы.
Во II стадии выявляются типичные изменения в субхондральном отделе эпифизов: краевые узоры, образование одиночных, овальной формы и мелкоячеистых деструкций и кист. Более выражен остеопороз, суставная щель сохранена, но может быть умеренно суженной, отмечается характерное изменение надколенника - квадратная форма его нижнего полюса и увеличение передне-заднего размера. Во П стадии функция сустава может быть умеренно сниженной, что проявляется небольшим ограничением амплитуды движений в нем, нарушением походки, гипотрофией мышц.
В III стадии сустав резко увеличен, дефигурирован, часто неровен и бугрист на ощупь, контрастирует с образующими его частями конечности, где определяется выраженная гипотрофия мускулатуры. Подвижность пораженных суставов в большей или меньшей степени ограничена, что связано как с поражением самого сустава, так и с изменениями мышц и сухожилий, которые часто укорочены, что приводит к развитию "конской стопы" и других нарушений, ограничивающих функцию конечности. Рентгенологически суставы утолщены, резко деформированы, суставные поверхности уплощены, эпифизы расширены за счет гиперостозов, диафизы уменьшены, суставная щель сужена. Выражен остеопороз, легко возникают внутрисуставные переломы. В бедренной кости отмечается типичное для гемофилии кратеро- или туннелеподобное разрушение костного вещества в области межмыщелковой ямки. Возможны различного рода подвывихи и смещения костей. Внутрисуставные хрящи разрушены.
В IVстадии функция сустава почти полностью утрачивается, суставная щель сужена, плохо контурируется на рентгенограмме, часто заращена соединительной тканью. С возрастом тяжесть и распространенность суставного поражения прогрессирует. Интенсивность прогрессирования поражения сустава зависит от частоты острых гемартрозов, своевременности и полноценности их лечения.
При гемофилии чрезвычайно тяжелы и опасны обширные и напряженные подкожные, межмышечные, субфасциальные и забрюшинные гематомы. Постепенно увеличиваясь они могут достигать огромных размеров, содержать от 0,5 до 3 литров крови, распространяться по межмышечным и другим пространствам далеко от места возникновения. Такие гематомы болезненны, иногда флюктуируют, вызывают анемию, повышение температуры тела до 38-38,5 °С и умеренный нейтрофильный лейкоцитоз, в связи с чем иногда ошибочно принимаются за флегмону и вскрываются, что еще более осложняет дальнейшее ведение больного. Вместе с тем гематомы иногда действительно инфицируются и нагнаиваются. При гематомах икроножных мышц мышца склерозируется и атрофируется, отмечается остеоидное изменение мышцы, образуется “конская стопа”, требующая ортопедического вмешательства. Кровоизлияние в брыжейку, сальник, стенку кишечника способствует компрессии кишечника, сосудов, возможно развитие кишечной непроходимости, гангрены. Опасность гематом состоит и в том, что они давят на окружающие ткани и питающие их сосуды, вызывают их некротизирование, сдавливая нервные стволы вызывают параличи, контрактуры, нарушение чувствительности, быстро прогрессирующую атрофию мышц. Кровоизлияние в подвздошно-поясничную мышцу характеризуется болями в животе, ригидностью мышц передней брюшной стенки, сгибательной контрактурой бедра. В области пораженной мышцы пальпируется плотное болезненное образование. УЗИ позволяет четко определелить место кровоизлияния.
Особого упоминания заслуживают обширные кровоизлияния в мягкие ткани подчелюстной области шеи, зева и глотки, вызывающие стенозирование верхних дыхательных путей и асфиксию.
Ушиб глаза может вызвать ретроорбитальное кровотечение, экзофтальм, сдавление нерва и слепоту. Спинальные гематомы редки, но очень опасны для жизни. При кровоизлиянии в спинной мозг развивается тетраплегия, однако, покой и лечение способствуют полному восстановлению функций конечностей, а если кровоизлияние организуется в спинномозговом канале и длительно сдавливает спинной мозг, долго держатся явления тетра- и параплегии с парезом тазовых органов, анестезий кожи, атрофией мышц. Кровоизлияния в головной и спинной мозг и их оболочки при гемофилии почти всегда связаны либо с травмами, либо с приемом анальгетиков, нарушающих функцию тромбоцитов. Между моментом получения травмы и развитием кровоизлияния может быть светлый промежуток продолжительностью от 1-2 ч до суток.
Кровоизлияние в головной мозг может произойти без предшествующей травмы головы. Вслед за нарастающим беспокойством или заторможенностью, рвотой и жалобами на головную боль появляются стволовые симптомы: горизонтальный и вертикальный нистагм, анизокария, расстройство ритма дыхания и сердечных сокращений. Расширенный, слабо реагирующий на свет зрачок - ранний признак внутричерепной гематомы.
Характерная черта гемофилии - длительные, повторно возобновляющиеся, опасные для жизни кровотечения при травмах и операциях. Кожные кровотечения, вызываемые небольшими травмами, порезами, царапинами могут длиться до 16 дней и более и привести к большим кровопотерям. Кровотечением из слизистых оболочек страдает 3/4 больных гемофилией. Наиболее часты кровотечения из носа и десен.
Серьезную терапевтическую проблему при гемофилии создают обильные и упорные почечные кровотечения, наблюдающиеся у 14-30% больных, в основном, старше 5 лет. Они намного труднее поддаются терапии, чем геморрагии других локализаций. Гематурия часто сопровождается у больных дизурическими явлениями, болями в поясничной области и приступами почечной колики, обусловленными образованием сгустков крови в мочевыводящих путях. Особенно интенсивными и выраженными эти явления становятся при лечении больных, когда временно восстанавливается нормальный гемостаз. Почечные кровотечения склонны к рецидивированию. При частых рецидивах возможно присоединение вторичной инфекции почек, редко исход в амилоидоз, ХПН. Кровоизлияния могут возникать как спонтанно, так и в связи с травмами поясничной области, сопутствующим пиелонефритом, приемом нестероидных противовоспалительных средств.
Желудочно-кишечные кровотечения могут быть спонтанными или обусловлены приемом препаратов, вызывающих эрозирование слизистой оболочки. Источником кровотечения служат язвы желудка и двенадцатиперстной кишки. Кровоизлияния в органы брюшной полости имитируют различные острые хирургические заболевания., реже травмы. В клинике быстрая анемизация, кома. В диагностике помогают УЗИ, КТ, иногда эндоскопические методы.
Единственным ориентиром в подобных ситуациях может быть эффективность интенсивной заместительной терапии в течение первых часов после начала абдоминальной катастрофы.
Осложнения. По мере увеличения продолжительности жизни больных гемофилией в результате интенсивной заместительно-трансфузионной терапии отмечается нарастание частоты осложнений этого заболевания.
Ингибиторная форма гемофилии развивается при появлении в крови больных в высоких титрах иммунных ингибиторов фактора VIII или фактора IX. Частота ингибиторной формы, по данным различных авторов, от 5 до 15%.
Гематомы у больных гемофилией могут подвергаться оссификации, приводить к деструкции костей и хрящей, возникновению внутри- и внесуставных переломов, чему способствует также выраженный остеопороз.
Часты при гемофилии и осложнения со стороны опорнодвигательного аппарата (подвывихи, контрактуры, укорочения ахиллова сухожилия и т.д.).
Кровоизлияния в головной и реже спинной мозг у больных гемофилией относительно редки, но крайне опасны и дают высокую летальность.
Большая группа осложнений при гемофилии связана с компрессией гематомами полых органов с их стенозированием (стенозы гортани, трахеи, кровеносных сосудов, кишечника и др.), а также нервных стволов, что ведет к самой разнообразной симптоматике - от асфиксии и парезов до кишечной непроходимости. Возможно инфицирование гематомы, гнойные артриты; развития псевдоопухоли. Трансфузионная терапия ведет к сенсибилизации больных, образованию циркулирующих иммунных комплексов и развитию гемолитической анемии, а также создает высокий риск заражения больных вирусами гепатита, приобретенного иммунного дефицита.
Лабораторные исследования.
Для ориентировочной диагностики решающее значение имеет выявление гипокоагуляции в таких общих пробах, как активированное парциальное тромбопластиновое время (АПТВ) и ауто-коагуляционный тест (АКТ). Показатели тромбинового и протромбинового времени остаются нормальными. При тяжелых и среднетяжелых формах гемофилии обнаруживается выраженное удлинение общего времени свертывания плазмы, снижение потребления протромбина. Однако, эти методы не позволяют диагностировать менее глубокие нарушения гемостаза.
Различают специфические и неспецифические тесты, позволяющие выявить основное звено в нарушении гемокоагуляции.
I. Неспецифические тесты.
1. Время свертывания плазмы по Ли-Уайту (N - 5-8 мин) у больных гемофилией удлинено. Тест малоинформативен, так как изменения регистрируются только при грубых нарушениях гемостаза.
2. Активированное парциальное тромбопластиновое время (N - 30-40 сек), резко удлинено. Это - лучший тест для выявления скрытых дефектов коагуляции, отражает недостаточность внутреннего механизма свертывания.
II. Специфические тесты.
1. Определение уровня факторов IX или VIII.
2. Альтернативно, при отсутствии возможности прямого определения уровня фактора, можно использовать аутокоагуляционный тест (АКТ) в сочетании с коррегирующими тестами.
3. Идентификация вида гемофилии может быть выполнена и тестами смешивания: к плазме исследуемого больного последовательно в разных пробирках добавляют образцы плазм больных с заведомо известной формой гемофилии, с почти нулевым содержанием факторов. Если смешиваются плазмы с недостатком одного и того же фактора свертывания, то коррекции нарушений свертываемости не происходит. При смешивании же плазмы с различными нарушениями происходит взаимная компенсация дефектов и полная нормализация свертывания. Следовательно, форма гемофилии устанавливается на той плазме, которая не исправляет у исследуемого больного времени свертывания.
III. Группа крови, резус-фактор.
IV. По показаниям протромбин, фибриноген, рентгенография костно-суставной системы.
V. УЗИ почек, печени, селезенки.
VI. Рентгенография костно-суставной системы при имеющихся изменениях.
Антенатальная диагностика. При первичном обследовании должны быть учтены следующие важные моменты: выявление носителей (кондукторов) и пренатальная диагностика гемофилии. Среди известных методов выявления носителей простейшим является генеалогическое дерево и определение цветового зрения (гены VIII фактора и цветового зрения близко расположены, доказано его нарушение у больных гемофилией).
В экономически развитых странах используются:
- линейный анализ полиморфизма ДНК с изучением внутренних и внешних маркеров дефектности гена. Исследование сложное и дорогое;
- в семьях с одним больным мужчиной трудно определить статус носителя методом линейного анализа ДНК, поэтому предложена модель на основе рекомендации в 22 интроне методом блот-анализа.
Пренатальная диагностика включает:
- определение антигена факторов VIII и IX и их свертывающей активности в крови плода. Кариотип плода и ДНК могут быть получены из плацентарной ткани путем пункции хориона с 10-й недели гестации. Риск потери плода приблизительно 1%. Длительность исследования несколько дней;
- ранний амниоцентез (11-14 нед гестации) может быть высоко информативным, но количество получаемых клеток невелико и требуется 2-3 нед на исследование;
- для желающих избежать инвазивную процедуру может быть проведена сонография в 15-16 нед гестации, если плод мужского пола, принимается дальнейшее решение;
- пункция пуповины - после 17 нед гестации. Риск прерывания беременности приблизительно 1%. Определение антигенов VIII и IX факторов, их свертывающей активности. Это исследование предпочтительнее в конце беременности, если есть информация о носительстве, а плод мужского пола, чтобы в родах оказать помощь новорожденному из-за опасности внутричерепного кровоизлияния;
- тестирование всех семей, где есть больные гемофилией, является перспективным, но экономически очень нагрузочным.
Дифференциальный диагноз. Гемофилия должна быть заподозрена во всех случаях, когда имеется гематомный тип кровоточивости с поражением опорно-двигательного аппарата, а также при длительных поздних кровотечениях после травм и хирургических вмешательств. Большое значение имеет семейный анамнез, пол ребенка. Гемофилию следует дифференцировать от болезни Виллебранда. Это заболевание обусловлено нарушением синтеза в эндотелии сосудов и поступлением в кровоток аутосомного компонента VIII фактора свертывания крови, фактора Виллебранда (VIII:ФВ), ответственного за адгезию тромбоцитов. Наследуется аутосомно - доминантно. Тип кровоточивости - смешанный (микроциркуляторно-гематомный). Клиническая картина характеризуется прежде всего кровоточивостью из слизистых оболочек, внутрикожными и подкожными кровоизлияниями. При тяжелой форме возможны кровоизлияния в суставы, но они редки и не заканчиваются развитием остеоартроза, как при гемофилии Характерно значительное удлинение времени кровотечения по Дюке, снижение ристоцин-агрегации тромбоцита, снижение активности фактора Виллебранда в плазме.
Лечение гемофилии.
Принципы терапии основаны на заместительном введении недостающего фактора. Дозы и длительность зависят от уровня VIII и IX факторов у больного, вида кровотечений, причины, вызвавшей кровотечение. Следует помнить, что фактор VIII лабилен и разрушается.
Лечение гемофилии А
Наиболее эффективные при гемофилии
1. Концентраты фактора VIII: Гемофил М, Иммунат, Коэнт ДВИ, Эмоклот, Когенэйт ФС и др. Флаконы от 300 до 1600 ME, а также рекомбинантные факторы (rVIII, rIX), препараты, полученные из плазмы животных, высокой степени очистки, безопасные по передаче вирусных и бактериальных инфекций.
2. Каждая единица фактора VIII на кг массы тела поднимает уровень VIII фактора в крови» на 2%. Период полураспада» 8-12 часов. Расчетную дозу необходимо соотнести с показателями фактора VIII в крови у пациента.
3. Расчет: масса пациента х на желаемый уровень фактора в % х 0,5.
4. Фактор вводить внутривенно, медленно, скорость 100 единиц в минуту. Концентрат фактора VIII 20-30 ЕД/кг повышает уровень фактора на 50%.
5. Всегда вводить весь объем флакона, даже если доза превышает расчетную. Преимущества фактора - концентрированы, очищены, не требуют особых условий для хранения, удобны в применении, низкий риск инфицирования.
При отсутствии перечисленных препаратов используется криопреципитат замороженный или лиофилизированный только при состояниях, угрожающих жизни больного.
Недостатки: менее очищенный, содержит фактор VIII, фактор Виллебранда, фибриноген, фактор XIII; не стандартный по активности; замороженный КП требует особых условий хранения (t - 20°С).
Дозировка: Доза зависит от тяжести кровотечения, исходного уровня VIII или IX фактора в крови.
Расчет: Д = (М х Уф): 1,3 (Д- доза единиц фактора, М - масса больного в кг, У - уровень фактора)
При нетяжелых и среднетяжелых кровотечениях:
КП = 1 пакет на 5 кг веса больного повышает уровень фактора в среднем на 50% от нормы.
СЗП. Недостатки: невозможность поднять уровень VIII (IX) фактора, более 20-25% от исходного уровня из-за опасности гемодинамической перегрузки; требует особых условий хранения, неудобен в использовании; сохраняется риск инфицирования; не стандартна по активности; выше риск сенсибилизации и развития ингибиторных форм гемофилии.
СЗП - суточная доза не должна быть более 30 мл/кг, разовая доза не более 15 мл/кг.
ВНИМАНИЕ:
- СЗП и КП должны вводиться с учетом группы крови;
- все препараты должны вводится внутривенно струйно;
- КП не следует использовать при гемофилии В;
- кратность (при необходимости повторных введений): гемофилия А 3-4 раза в сутки, гемофилия В 1 - 2 раза в сутки.
В. Лечение гемофилии В
1. Концентраты фактора IX: Иммунин, Аимафикс, Октанайн и др. (см. инструкцию к препаратам). Флаконы от 300 до 1200 ME.
2. Каждая единица фактора IX на кг массы повышает уровень фактора в плазме» на 1%.
3. Расчет: масса пациента х на желаемый уровень фактора в %.
4. Вводить внутривенно медленно,» 3 мл в минуту
5. СЗП не должна использоваться, если только нет угрозы жизни или если нет фактора IX (невозможно достичь при дозе 15 мл/кг уровня более 15%).
6. КП не вводить
7. Аминокапроновую кислоту не вводить.
Таблица 8

