




Вплив складу газового середовища на прикладі О2, Н2О, СО2 і SО2 і температури на корозію окремих металів показано на рис. 1.16.
Залізо виявляє досить високу корозію у всіх досліджених газових середовищах, що значно зростає в інтервалі 700-900 °С.
Хром має високу жаростійкість у всіх чотирьох атмосферах.
Нікель відносно стійкий в середовищі О2, Н2О і СО2, але сильно кородує в атмосфері SО2.
Кобальт найбільшу швидкість корозії має в середовищі SО2, що значно зростає при переході від температури 700 °С до 900 °С.
Мідь найбільш швидко кородує в атмосфері кисню, але стійка в середовищі SО2.
Прийнято вважати, що відмінності в швидкості корозії металів в різних газах значною мірою визначаються захисними властивостями утворених на металі плівок продуктів корозії.
1.10. Теорії жаростійкого легування
В багатьох хімічних процесах металеві конструкції і вироби експлуатуються в жорстких умовах, при підвищених температурах, великих тисках, високій агресивності середовища. Чисті метали, як правило, не є корозійностійкими і вимагають додаткових заходів захисту. Сучасна техніка протикорозійного захисту має ряд ефективних способів для збільшення стійкості металів і терміну їх експлуатації. Щодо умов газової корозії одним з найбільш часто використовуваних способів є жаростійке легування з метою одержання сплавів, що володіють підвищеною корозійною стійкістю.
В даний час існує три найбільш обґрунтовані теорії жаростійкого легування, які не суперечать, а швидше доповнюють одна одну.
Відповідно до першої теорії, розробленої Вагнером і Хауффе, невелика добавка легуючого елемента окислюється і, розчиняючись в оксиді основного металу, зменшує число дефектів у кристалічній решітці основного металу. Це призводить до впорядкування структури і знижує швидкість дифузії іонів в захисній плівці. Дана теорія має досить обмежену сферу застосування. Якщо швидкість окислення металу визначається не дифузією іонів або при легуванні в оксидній плівці утворюється нова фаза, то зазначені вище припущення теорії жаростійкого легування незастосовні.
Згідно другої теорії, розробленої А.А. Смирновим, Н.Д. Томашовим і ін., на поверхні металу утворюється захисний оксид легуючого елемента, який перешкоджає окисленню основного металу. Стосовно до цієї теорії легуючий компонент повинен володіти перерахованими основними властивостями:
| Збільшення маси, Кm+, г/(м3∙год.) | 
|
| 700 0С 900 0С |
Рисунок 1.15. Швидкість газової корозії деяких металів в атмосферах О2, Н2О, СО2 та SO2 за 24 години при 700 і 900 0С.
1. Оксид легуючого елемента повинен задовольняти умові суцільності, тобто відношення об’ємів оксиду і металу має бути більше одиниці: Vок / VMе > 1. У табл. 1.3 наведені узагальнені дані по деяких властивостях оксидів хрому, алюмінію і кремнію. Ці елементи є основними добавками для підвищення жаростійкості заліза. Як показують дані таблиці ця умова для вищеназваних елементів виконується.
2. Легуючий елемент повинен утворювати оксид з високим електричним опором. Великий омічний опір (низька електропровідність) є однією з основних умов для формування захисних властивостей плівки, тому що при цьому рух іонів у шарі оксиду ускладнюється.
3. Енергія утворення оксиду легуючого елемента повинна бути більше енергії утворення оксиду основного металу.
Ця умова забезпечує стійкість оксиду легуючого компонента в присутності основного металу. Оксид компонента добавки виявляється більш стійким, ніж оксид основного металу. Якщо ця умова не дотримується, то оксид легуючого елемента буде відновлюватися основним металом.
Наведені в таблиці 1.3 дані по теплотам утворення оксидів заліза, Аl2O3, Сг2О3 і SiO2 підтверджують сформульоване вище правило. Більше того, якщо спочатку утворюється змішаний оксид, то надалі відповідно до умов термодинамічної рівноваги, він переходить у чистий оксид легуючого компонента.
Наприклад, при окисленні заліза легованого алюмінієм, має місце утворення оксиду заліза FeО. Але далі можливий процес:
3FeO + 2Al = Al2O3 + 3Fe/
4. Розмір іонів легуючого елемента повинен бути менше за розмір іона основної металу (див. табл. 1.3). Це полегшує дифузію легуючого елемента до поверхні сплаву, на якому утворюється захисна плівка.
Таблиця 1.3
Деякі властивості оксидів елементів, що підвищують жаростійкість заліза
| Властивості | Оксид | ||||
| FеО | Fе2O3 | А12O3 | Сг2O3 | SiO2 | |
| Відношення Vок / VMе | - | 2,14 | 1,28 | 2,07 | 1,9 |
| Електропровідність, Ом–1∙см–1 при 1000 °С (порядок значения) | 102 | - | 10–7 | 10–1 | 10–6 |
| Іонний радіус еталлу в оксиді, А | 0,75 | - | 0,54 | 0,6 | 0,41 |
| Теплоти утворення: кДж/моль кДж/атом | 269,2 269,2 | 801.8 400,9 | 1674,7 837,4 | 1122,0 561,0 | 818,2 818,2 |
5. Оксиди легуючих компонентів повинні мати високі температури плавлення і сублімації та не утворювати низькоплавкої евтектики. Ця вимога забезпечує збереження оксиду при високих температурах у твердій фазі. Перехід оксиду в рідкий стан полегшив би протікання в ньому дифузійних процесів. Часткова сублімація оксиду збільшила б пористість плівки, що сприяє зниженню її захисних властивостей.
6. Легуючий компонент і основний метал повинні утворювати твердий розчин при даному складі сплаву. Тільки за цієї умови вдається забезпечити суцільну плівку оксиду легуючого компоненту по всій поверхні сплаву.
Ця теорія жаростійкого легування знаходиться в хорошій злагоді з цілим рядом практичних даних. Експериментально було встановлено наявність захисного шару оксиду, переважно утвореного легуючим компонентом сплаву (хрому або алюмінію) для ряду жаростійких залізних сплавів. На рис. 1.16 показано значне зниження швидкості окислення заліза від концентрації легуючої добавки - алюмінію.
| Відносна швидкість окислення k | 
|
| Вміст алюмінію, % |
Рисунок 1.16. Вплив легування заліза алюмінієм на відносну швидкість окислення на повітрі при 900 0С (швидкість окислення чистого заліза (k) прийнята за 100%).
2 ЕЛЕКТРОХІМІЧНА КОРОЗІЯ
2.l Виникнення електродного потенціалу
Електрохімічна корозія металів представляє собою самовільне руйнування металу внаслідок електрохімічного взаємодії його з електролітом. Це гетерогенна реакція, що протікає на поверхні металу. Причиною електрохімічної корозії є термодинамічна нестійкість металу в даних корозійних умовах.
Для розуміння механізму електрохімічної корозії необхідно встановити, які процеси спостерігаються на межі метал - розчин. На межі розділення двох фаз при певних умовах може виникнути різниця потенціалів або, як прийнято говорити, стрибок потенціалу(наприклад електродний потенціал - на межі метал - розчин; контактний потенціал - на межі двох різних металів; контактний потенціал другого роду - на межі метал - газ; дифузійний потенціал - на кордоні двох розчинів), що мають різну концентрацію розчиненої речовини, та ін.
Стрибок потенціалу між двома фазами визначається переходом заряджених часток з однієї фази в іншу або вибірковою адсорбцією заряджених або полярних частинок однієї фази на поверхні іншої з утворенням подвійного електричного шару.
Розглянемо механізм виникнення електродного потенціалу. Електродом називається метал, занурений у розчин електроліту. Що ж відбувається на поверхні металу при зануренні його в розчин власних іонів?
У металі і в розчині є однакові іони - іони металу. У металі ці іони перебувають у вузлах кристалічної решітки і утримуються в ній завдяки енергії зв'язку іонів решітки. Щоб вивести іон з кристалічної решітки, необхідно затратити роботу, рівну енергії зв'язку іонів, яку також можна назвати роботою виходу іона з металу.
В розчині іони металу оточені полярними молекулами води, тобто перебувають у гідратованому стані. Щоб вивести іон металу з розчину необхідно виконати роботу, рівну енергії гідратації, тобто енергії зв'язку іона металу з молекулами води.

Рисунок 2.1. Схема зміни енергії при випаровуванні катіона металу у вакуум і при переході в розчин:
а - момент занурення металу в розчин його солі; б - момент встановлення рівноваги.
Встановлення електродного потенціалу на металі залежить від співвідношення енергії кристалічної решітки та енергії гідратації іонів. Знаходяться на поверхні металу катіони мають запас потенційної енергії, що відповідає значенню енергії в точці 1 (нижня) на рис. 2.1. Відрив катіона від поверхні металу з переходом у вакуум вимагає значної енергії (пунктирна крива 1-1), відповідної енергії випаровування Qвип. Полярні молекули води (або іншого розчинника), орієнтуючись навколо поверхневих катіонів металу, полегшують перехід катіонів у розчин із звільненням енергії гідратації, тому що рівень енергії гідратованого іону нижче, ніж катіона у вакуумі, на величину Qгідр. Потенційна енергія катіонів, що знаходяться в розчині, в межах подвійного електричного шару відповідає точці 2. Для переходу в розчин поверхневий катіон металу повинен подолати лише енергетичний бар'єр Qа. Різниця рівнів потенційних енергій в точках 1 і 2, що дорівнює А, відповідає роботі процесу переходу іонів металу в розчин. Для переходу з розчину в метал гідратований катіон повинен подолати енергетичний бар'єр Qк. Відповідно до теорії О.М.Фрумкіна, при взаємодії металу і розчину протікають два сполучених процеси:
1. Перехід іонів з металу в розчин з утворенням гідратованих іонів (анодний процес):
Me + mН2O = Меn+ · mН2O + ne.
Швидкість цього процесу, виміряна числом іонів, що переходять з однієї фази в іншу через одиницю поверхні за одиницю часу, може бути виражена через щільність струму ir.
2. Перехід іонів з розчину з виділенням їх на поверхні металу у вигляді нейтральних атомів, що входять до складу кристалічної решітки металу (катодний процес):
Меn+ · mН2O + ne = Ме + mН2O.
Швидкість катодного процесу виражається через відповідну щільність струму is. Який з цих процесів переважає, визначається рівнем потенційної енергії катіонів у вузлах кристалічної решітки металу UМе і в розчині Uр.
Якщо UМе > Uр, то переважає анодний процес, сумарна швидкість якого iа =ir – is. Розчин отримує надлишковий позитивний заряд у вигляді катіонів металу, а поверхня металу набуває надлишковий негативний заряд за рахунок надлишку електронів. Перехід частини катіонів у розчин супроводжується зниженням середньої потенційної енергії поверхневих катіонів (точка 1 переміщається вниз до точки 1'), появою на металевій поверхні надлишкових негативних зарядів і підвищенням енергетичного бар'єру Qа. Підвищення концентрації іонів біля поверхні металу супроводжується зростанням потенційної енергії (точка 2 переміщається вгору до точки 2'), придбанням розчином надлишкового позитивного заряду і зниженням енергетичного бар'єру Qк. По мірі збільшення концентрації катіонів біля поверхні, із зростанням величини заряду розчину і металу, протікання прямого процесу ускладнюється і полегшується перебіг зворотного процесу, тобто перехід іонів металу з розчину в кристалічну решітку.
Коли енергетичні рівні іонів на поверхні металу і в розчині стають однаковими, тобто UМе = Uр, встановлюється динамічна рівновага. Рівновага характеризується тим, що Qк = Qа = Qо і швидкості анодного і катодного процесів є рівними: ir = is = iо, де iо - щільність струму обміну.
Внаслідок електростатичного притягання катіонів та надлишкових електронів на поверхні металу іони металу не можуть віддалитися вглиб розчину, а перебувають біля поверхні. Утворюється подвійний електричний шар (рис.2.2, а).Різниця електричних потенціалів, що виникає на межі метал - розчин, внаслідок надлишкових зарядів, називається електродним потенціалом.
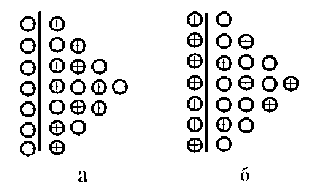
Рисунок 2.2. Подвійний електричний шар системи: а – Zn/ZnSO4; б – Cu/CuSO4.
Якщо UМе ˂ Uр, то потенційна енергія катіона в металі менше потенційної енергії гідратованого іону. Тоді в початковий момент відбувається не розчинення, а осадження металу з розчину, сумарна швидкість якого ік = is – ir. При цьому поверхня металу набуває надлишковий позитивний заряд катіонів, a надлишок аніонів що залишився в приелектродному просторі надає йому негативний заряд. Позитивний заряд поверхні металу ускладнює подальше осадження катіонів і полегшує зворотний процес - перехід іонів металу з решітки в розчин. В результаті в системі встановлюється динамічна рівновага і виникає подвійний електричний шар з протилежним розподілом зарядів, тобто поверхня металу заряджається позитивно, а в розчині у поверхні металу утворюється надлишок аніонів (рис.2.2, б).Таким чином, незалежно від енергетичних співвідношень, настає динамічна рівновага між металом і розчином його солі, яка характеризується певною за величиною і знаком рівноважним потенціалом.
2.2 Рівноважні і не рівноважні електродні потенціали
При зануренні металів в розчин будь-якого електроліту виникає електродний потенціал. Якщо у встановленні потенціалу приймають участь тільки власні іони металу (заряди переносяться тільки ними), то такий потенціал називають рівноважним, або оборотним (рис. 2.3, а). Величина рівноважного електродного потенціалу залежить від природи металу і розчинника, температури, активності іонів металу в розчині. Рівноважні потенціали металів, визначені для активності іонів металу в розчині, що дорівнює одиниці, при температурі 25 °С, називають стандартними електродними потенціалами. Стандартні електродні потенціали можна розрахувати по зміні ізобарно - ізотермічних потенціалів електродних процесів, віднесених до 1 моль металу і виражених у вольтах:
ΔG = – nFE; E = – (ΔG/nF). (2.1)
За відомим значенням енергії Гіббса реакції (ΔG) можна розрахувати величину електродного потенціалу. Рівняння (2.1) показує перетворення хімічної енергії в електричну та назад.
Значення стандартних електродних потенціалів для деяких металів приведені в табл.2.1.
Ряд металів, розташований за зростанням позитивних значень стандартних електродних потенціалів, називається рядом напруг.
Положення металу в ряду визначає його хімічну активність, окисні і відновні властивості. Чим більш негативне значення потенціалу має метал, тим більшою мірою зростає його здатність до окислення.
Таблиця 2.1
| Електрод | K+ | K↔ | Al3+|Al↔ | Ti2+|Ti↔ | Mn2+|Mn↔ | Zn2+|Zn↔ | ↔Cr3+|Cr |
| E0, В | - 2,925 | - 1,66 | - 1,63 | - 1,18 | - 0,762 | - 0,74 |
| Електрод | Fe2+|Fe | 2H+|H2↔ | Cu2+|Cu | Hg2+|Hg | Pd2+|Pd | Pt2+|Pt↔ |
| E0, В | - 0,447 | 0,000 | + 0,337 | + 0,799 | + 0,987 | + 1,19 |
Залежність рівноважного електродного потенціалу від активності іонів металу в розчині і температури визначається формулою Нернста:
Е + Е0 + (RT / nF)∙lnaМеn+, (2.2)
де E0 - стандартний електродний потенціал металу, В, Т - температура вимірювання потенціалу, К; n - ступінь окислення металу; F - число Фарадея, 96500 Кл; R - універсальна газова стала, 8,31 Дж / К; а - активність іонів металу в розчині, г-іон / л.
Підставивши значення всіх констант (при Т = 298 К) в формулу 2.2, одержимо:
E + E0 (0,059 / n)lgaМеn+ (2.3)
Приклад. Визначити потенціал мідного електроду в розчині CuSO4, якщо Cu2+ = 0,001 г-іон / л, E0 = + 0,337 В: E = 0,337 + (0,059 / 2) lg 10-3 = 0,337 - 0,088 = = + 0,249 (В).
У багатьох практичних випадках на металах (мідь, ртуть, срібло) встановлюються рівноважні, або оборотні, потенціали. Абсолютні значення стандартних потенціалів визначити експериментально і обчислити теоретично не представляється можливим. У зв'язку з цим їх визначають по відношенню до стандартного водневого електрода, потенціал якого умовно прийнятий рівним нулю.
Якщо у встановленні електродного потенціалу приймають участь не тільки власні іони металу, але й інші іони і атоми, то виникають не рівноважні, чи необоротні, потенціали. Умовою утворення не рівноважного потенціалу є рівність швидкостей переносу зарядів у прямому і зворотному напрямках, тобто баланс заряду, але баланс маси при цьому не дотримується, так як у передачі зарядів беруть участь різні часточки (рис. 2.3, б).

Рисунок 2.3. Схема встановлення потенціалів: а - рівноважного; б - не рівноважного.
Стале в часі значення незворотного електродного потенціалу металу, відповідне рівності сум швидкостей анодних і катодних процесів, називають стаціонарним потенціалом металу. Метал переважно розчиняється, а баланс зарядів, які переносяться у зворотному напрямку, компенсується іонами металу та іншими частками, наприклад переходом Н+ з розчину в газову фазу.
Таким чином, при встановленні на металі незворотного електродного потенціалу може відбуватися електрохімічне розчинення металу:
Me + mH2O → Men+1∙mH2O + ne (анодний процес)
і відновлення будь-якого деполяризатора (іона або молекули), що знаходиться в розчині, наприклад, іонів водню:
H+H2O + e → H + H2O (катодний процес).
До необоротних електродних потенціалів відносяться потенціали багатьох металів у розчинах власних іонів (нікель, залізо, хром, титан та ін.) Для не рівноважних потенціалів формула Нернста непридатна, тому що електродний потенціал визначається декількома паралельними реакціями.
Величини необоротних електродних потенціалів металів залежать як від внутрішніх чинників, пов'язаних з природою металу, так і від зовнішніх, пов'язаних зі складом електроліту і фізичними умовами. До внутрішніх факторів належать: фізико-хімічний стан і структура металу, стан поверхні, наявність механічних деформацій і напружень та ін. Зовнішні чинники - це хімічна природа розчинника, природа і концентрація розчинених газів, температура, тиск, перемішування розчину та інші. Не рівноважні електродні потенціали визначаються тільки дослідним шляхом. У табл. 2.2 наведено дослідні значення стаціонарних потенціалів ряду металів в нейтральних середовищах.
Таблиця 2.2
| Потенціал | Al | Zn | Cr | Fe | Ni | Cu |
| E0, В | - 1,66 | - 0,762 | - 0,74 | - 0,44 | - 0,23 | + 0,337 |
| Eстац. 3% NaCl | - 0,63 | - 0,83 | + 0,23 | - 0,50 | - 0,02 | + 0,05 |
| Eстац. 1% Na2SO4 | - 0,47 | - 0,81 | - | - 0,5 | + 0,035 | + 0,24 |
Для вирішення питання, чи є потенціал даного металу в будь-якому електроліті оборотним або незворотнім, слід зіставити теоретичне значення, розраховане за рівнянням (2.1), і значення електродного потенціалу металу, отримане дослідним шляхом.
2.3 Будова подвійного електричного шару
Як було показано раніше, на межі метал - розчин утворюється подвійний електричний шар. Іони, що зібралися біля поверхні металу при встановленні рівноваги, не можуть покинути приелектродний шар і віддалитися від нього углиб розчину. Цьому перешкоджає електростатичне притягання між іонами і надлишковим зарядом на поверхні металу. Однак подвійний електричний шар може утворитися і без переходу заряджених частинок з фази у фазу. У цьому випадку утворення подвійного шару можливо за рахунок вибіркової адсорбції іонів однієї фази на поверхні іншої, наприклад специфічна адсорбція аніонів хлору з водного розчину солі на поверхні будь-якого інертного металу. Це призводить до появи в прикатодному шарі надлишкового негативного заряду і позитивного заряду в прилеглому шарі розчину.
а б
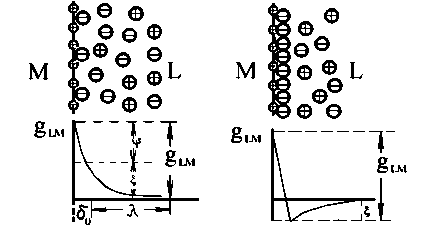
Рисунок 2.4. Будова подвійного електричного шару: а - при відсутності в розчині поверхнево-активних речовин; б - при їх наявності.
Будова подвійного шару і зміна потенціалу із збільшенням відстані від поверхні металу для розчинів, що не містять поверхнево-активних речовин, показані на рис. 2.4 а; для розчинів, що містять поверхнево-активні аніони - на рис. 2.4, б. Відповідно до теорії Штерна, подвійний електричний шар поділяється нa щільну частину δ0, товщина якої дорівнює середньому радіусу іонів електроліту, і дифузійну частину, де концентрація іонів поступово падає, досягаючи концентрації, властивої цьому розчину в цілому.
Величина електродного потенціалу складається з потенціалів щільної ψψ і дифузної ψζ' частин подвійного шару: gLM = ψ ψψ + ζ'ψ'. Загальна товщина подвійного шару складається з товщини, приблизно рівною радіусу сольватованого іону (щільна частина подвійного шару), і товщини дифузійної частини подвійного шару: μ = δ0 + λ.
Товщина дифузійної частини подвійного шару залежить від природи і особливо від концентрації розчину. У чистій воді товщина дифузної частини зменшується, а з підвищенням температури, внаслідок теплового руху розчину - збільшується.
За теорією Штерна, в щільній частині подвійного шару іони утримуються не тільки електростатичними силами, але і силами специфічної адсорбції, тобто силами некулонівского походження. Тому в розчинах, що містять поверхнево - активні іони, їх число в щільній частини подвійного шару може бути не еквівалентним заряду поверхні металу, а перевершувати його на деяку величину, що залежить від властивостей іонів і заряду металу.
Таким чином, слід розрізняти дві моделі подвійного електричного шару, одна з яких належить до розчинів, що не містять поверхнево активних речовин (рис.2.4, а), інша - до розчинів, що містить специфічно адсорбовані іони (рис.2.4, б).
2.4. Потенціал нульового заряду
Електрод з аніонним подвійним електричним шаром має позитивний потенціал щодо розчину (рис.2.5, а).

а б в
Рисунок 2.5. Схема перезарядки поверхні металу при катодній поляризації:
а - позитивний заряд; б - відсутність заряду; в - негативний заряд.
Якщо такий електрод піддати катодній поляризації, тобто послати на нього від зовнішнього джерела струму негативні заряди (електрони), то можна досягти зникнення на його поверхні надлишку позитивних зарядів, при цьому зникне і аніонний подвійний електричний шар (рис. 2.5, б). Потенціал, при якому поверхня металу не заряджена (відсутній іонний подвійний електричний шар), називають, по А. Н. Фрумкіна, потенціалом нульового заряду Eq = 0. При подальшій катодній поляризації металу відбувається перезарядка його поверхні з утворенням відповідного катодного електричного шару (рис. 2.5, в).
Л. І.Антропов запропонував замість загального поняття «потенціал нульового заряду» два нові терміна: потенціал незарядженої поверхні Eq = 0 і нульова точка EN. Потенціал незарядженої поверхні для даного металу і розчинника змінюється в широких межах залежно від природи і концентрації речовин, присутніх у розчині. Нульова точка - значення потенціалу незарядженої поверхні в розчині, не містить ніяких поверхнево - активних частинок. Це значення є константою, характерною для даного металу і даного розчинника. Нульова точка і потенціал незарядженої поверхні знаходяться між собою приблизно в такому ж співвідношенні, як стандартний і рівноважний потенціали електроду.
Джерелом ЕРС між металами при потенціалах нульових зарядів, з теорії А. М. Фрумкіна, може бути контактна різниця потенціалів, а також адсорбція іонів і полярних молекул. Різниця потенціалів нульових зарядів двох металів повинна бути приблизно рівною контактному потенціалу між ними (так званого контактного потенціалу Вольта):
ENMe1 − ENMe2 = φ1,2 = − (А1е−А2е)/F, (2.4)
де φ1, 2 - контактний потенціал Вольта між металами 1 і 2, В; Aе1, Aе2 - робота виходу електрона з металів 1 і 2, еВ.
Л.І. Антропов, прийнявши в якості другого еталонного металу ртуть, для якої величина роботи виходу електрона з металу і потенціал нульового заряду визначені AеHg = 4,52 еВ, EHgN = -0,19 В, одержав рівняння для розрахунку потенціалу нульового заряду будь-якого металу, якщо для нього відома робота виходу електрона:
ENMe1 = ENMe2 − (A1e − A2e)/F = A1e/F − 4,52 − 0,19 ≈ A1e/F − 4,71 (2.5)
Ця залежність наведена на рис.2.6 для ряду металів, з якої видно, що відхилення дослідних точок від теоретичної прямої в більшості випадків незначні.

Рисунок 2.6. Зв'язок між потенціалом нульового заряду і роботою виходу електрона у водних розчинах.
Л.І. Антроповим запропонована наведена φ-шкала, в якій за початок відліку прийнята нульова точка. Потенціал φ у наведеній шкалою визначається як різниця між потенціалом електрода EМе в даних умовах і його нульовою точкою:
Φ φ = EМе –EN. (2.6)
Потенціал за φ - шкалою дає характеристику заряду поверхні металу, по якому можна визначити, які поверхнево-активні речовини (катіонні, аніонні або нейтральні) можуть адсорбуватися на поверхні металу в даних умовах. При φ > 0 на поверхні металу переважає адсорбція негативно заряджених часток (аніонів), при φ ˂ 0 - позитивно заряджених часток (катіонів), і при φ ≈ = 0 - молекулярних часток. Адсорбція різних речовин на поверхні металу може дуже сильно змінити швидкість його корозії.
2.5 Термодинаміка корозійних електрохімічних процесів
Основною причиною корозії металів є їх термодинамічна нестійкість. Прагнення металів переходити з металевого стану в іонний (тобто розчинятися) для різних металів є неоднаковим і найбільш точно може бути охарактеризовано зміною вільної енергії при протіканні відповідної реакції окислення в даному середовищі. Відомо, що при самовільному процесі вільна енергія може тільки зменшуватися.

