




Недосконалий остеогенез характеризується структурними дефектами колагенових генів, при яких заміщується гліцин аргініном в 391 положенні. Ця заміна супроводжується підвищенням ферментної активності в сусідніх регіонах α -1 ланцюга, внаслідок чого порушується потрійна структура колагену і аномалій колаген не включається в „нерозчинну” фракцію кісткового колагену.
Клінічними ознаками недосконалого остеогенезу являється (рис. 1,2,3):
· підвищена ламкість довгих трубчастих кісток, ребер та ключиць
· голубі склери
· отосклероз
· укорочення, викривлення кісток, утворення несправжніх суглобів
· кифосколіоз
· деформації грудної клітини
· „риб’ячі” хребці
· жовто-коричневі зуби
· кили
гомоцистинурія
Гомоцистинурія (ГЦУ) була вперше описана в 1962 р. Джеррітсеном, Карсоном і Нейлом.
ГЦУ – генетично детерміноване порушення обміну амінокислоти метіоніну, що обумовлено блоком ферменту бета-цистатіонин-синтази. По колишнім даним частота ГЦУ 1:50000 – 1:250000. Зараз відомо, що помітна частина населення – 10%, має підвищену потребу у фолатах, тому що вона є носієм частого генетичного варіанта гена, що має відношення до метаболізму. Гомозиготність по мутації 677С (Т в локусі 5,10 – метілентетрагідрофолатредуктази (МТГФР), фолатнезалежного важливого ферменту в метаболізмі гомоцистеина, може робити фермент термолабільним і менш активним і асоціює зі збільшеним рівнем у плазмі крові загального гомоцистеїну. На жаль, частота мутантного МТГФР і його зв'язок з пороками розвитку в різних популяціях залишаються невідомими. Успадковується по аутосомно-рецесивному типі. Ген ГЦУ картований на довгому плечі 21 хромосоми (21 q 21-q22.1).
Таблиця1.
Дефіцит цистатіонін-β-сінтази
| Система | Симптоми/маркери | Підлітковий період | Дорослі |
| Незвичайні клінічні знахідки | Ектопія кришталику Затримка розвитку Тромбоемболічні епізоди Остеопороз | ± ± ± ± | ± ± ± ± |
| Специфічні лабораторні показники | Метіонін (П) Гомоцист(е)ін, вільний/загальний (П) Гомоцистеін-цистеін (П) Цистін (П) Гомоцистін (С) Гомоцистеін-цистеін (С) Цианід-нітропруссидний тест (М) | ↑ ↑ ↑ ↓ ↑ ↑ + | ↑ ↑ ↑ ↓ ↑ ↑ + |
| Центральна нервова система | Затримка розвитку Психіатричні симптоми Судоми Інфаркти | ± ± ± ± | ± ± ± ± |
| Очі | Міопія Ектопія кришталика | ± ± | ± ± |
| Скелетна система | Сколіоз Арахнодактилія-"марфаноїдні риси" Деформація грудини Вальгусна деформація колін Остеопороз | ± ± ± ± ± | ± ± ± ± ± |
| Судинна система | Окклюзії | ± | ± |
| Дерматологічні прояви | Сип | ± | ± |
П- плазма;
С- сеча.
Високий рівень метіоніну, зниження рівня цистину і поява гомоцистина – характерні біохімічні зрушення при цій патології. Той факт, що в гетерозигот рівень ферменту складає 25-30% нормального рівня пояснює, чому клінічно значимими є не тільки гомо-, але і гетерозиготні стани, тому те і виділені гетеро- і гомозиготні форми ГЦУ.
Дія, що ушкоджує, виявляється і при впливі на інтиму артерій середнього і великого калібру з наступною агрегацією на ній тромбоцитів у життєво важливих органах: у бруньках, селезінці, слизуватій оболонці шлунка, кістковій системі, судинах. Використання антикоагулянтів запобігає тромбоутворення, але не відновлює ушкоджену інтиму судин. Гомоцистин активує також фактор Хагемана, осідає в патологічно зміненій стінці судин і створює умови для утворення тромбів. Структурні зміни кісти при ГЦУ характеризуються остеопоpозом, викликаним гипепродукцією дерматансульфатів. При ГЦУ відзначається жирове переродження і білкова дистрофія печінки.
Патологія очей при ГЦУ характеризується дегенеративною поразкою в кругових волокнах кришталиків та циліарних тілах. При ГЦУ відзначаються виражені зміни обміну сполучної тканини, що носять вторинний характер, при цьому гомоцистин зв'язується з альдегідами – похідними лізину і перешкоджає утворенню поперечних зв'язків коллагена. Це знаходить своє вираження в збільшенні екстрактивності коллагену шкіри хворих, у підвищенні ниркової екскреції оксипроліну і глікозаміногликанів (ГАГ).
При ГЦУ переважає дерматанподібні фракції ГАГ, що являє собою компенсаторну реакцію організму, тому що дерматансульфат володіє антикоагулянтною активністю. Гомоцистин, у свою чергу підвищує активність синтезу сульфатированих ГАГ.
Для ГЦУ характерні: розумова відсталість, осередкова неврологічна симптоматика, ектопія кришталиків, кістякові деформації, тромбоемболія і кардіоваскулярна патологія. Ріст хворих – високий, астенічне статура, аранхнодактілія кистей і стіп, воронкоподібна чи килевидна деформація грудної клітки, кифосколіоз. Остеопороз кіст приводить до повторюваного переломам. Остеопороз при ГЦУ викликаний тим, що надлишок гомоцистеина перешкоджає нормальному синтезу поперечних зв'язків коллагену. Надлишок гормону росту при ГЦУ приводить до надлишкового росту кіст.
Патологія очей, виявляється подвивихом кришталика, глаукомою, що виникає через зсув кришталика в передню камеру чи ока, як наслідок, дегенеративних змін у тканинах передньої камери.
Патологія серцево-судинної системи при ГЦУ характеризуються тромбоемболією в артеріальних судинах різного калібру, порушенням обмінних процесів у міокарді.
Порушення нервової системи характеризуються спастичними паралічами, парезами, розумовою відсталістю через тромбози судин мозку.
Гіпопігментація є характерною рисою гомоцистинурії, що зникає при лікуванні пірідоксином. У деяких випадках після початку лікування пірідоксином волосся сутеніють чи виростають нові темні, з чіткою демаркацією між старим світлим та новим темним волоссям (Рис. 4).
У родинах з раннім захворюванням коронарних артерій 12 – 14 % мають гіпергомоцистеїнемію. Не менш яскраво просліджується взаємозв'язок гіпергомоцистеїнемії в родинах з венозними тромбоемболіями.
Атеросклероз, викликаний гомоцистеином, можна пояснити тим, що гомоцистеин сприяє росту гладком’язових кліток судин і придушує ріст ендотеліальних. Приблизно 30 % з передчасної окклюзивною хворобою артерій є гетерозиготами ГЦУ з гіпергомоцистеїнемієй. Виявлено, що високий рівень гомоцистеина в плазмі є чинником ризику тромбозу глибоких вен у звичайній популяції. Хворі з конкурентною гомоцистеїнурією, обумовленою недостатністю цистатіонин – бета-синтетази, мають збільшений ризик тромбозу, коли в них є присутнім мутація фактора 5 Лейдена. При цьому лікування варфарином із дня народження запобігає розвиток тромбозу. У жінок – гетерозигот по ГЦУ і по факторі 5 Лейдена відзначені спонтанні викидні й інфаркти плаценти.
ГЦУ генетично гетерогенна. В даний час виявлено 17 мутацій гена CBS, що приводять до клінічної маніфестації ГЦУ. Існує геногеографічна особливість мутацій і різний характер мутацій при різних клінічних проявах. Тому, ефективне лікування ГЦУ, адекватна реабілітація хворих і профілактика кардіоваскулярних ускладнень прямо зв'язані з точною діагностикою ГЦУ і її алельних варіантів. Разом з тим, приведені дані демонструють високу доцільність скринінгу немовляти на ГЦУ в тих регіонах, де її частота найбільш висока. Селективний скринінг на ГЦУ показаний:
- хворим з кістяковими аномаліями (марфоноїдний фенотип),
- с поразкою очей,
- кардіоваскулярною патологією,
- цереброваскулярною патологією,
- розумовою відсталістю,
- шизофренією,
- тромбозами і тромбоемболіями,
- гострими панкреатитами,
- у родинах, обтяжених атеросклерозом.
СИНДРОМ МАРФАНА (СМ)
СМ описаний у 1879 р. Вільямсом, а в 1896 – Марфаном як системне спадкоємне захворювання сполучної тканини (СТ) з аутосомно-домінантним типом спадкування і високої пенетрантністю мутантного гена. Популяційна частота 4:100000 чоловік.


Рисунок 4. Гомозиготна гомоцистинурія (дефіцит цистатіон-в-сінтетази)
Захворювання викликане мутацією одного гена, швидше за все, у локусі 15q21.1. Ген, що викликає це захворювання, може бути успадкований від батьків, що страждали цим захворюванням, чи виникнути вперше в результаті нової мутації. Біля 70% хворих з СМ мають хворих батьків і 30% є першими хворими в родині в результаті нових генних мутацій. Хворіють однаково часто чоловіки і жінки. Захворювання може носити генералізований характер унаслідок повної експресії патологічного гена, але може мати і стертий характер. Можуть бути різноманітні клінічні прояви в межах однієї родини.
У патогенезі СМ на перший план виступають поразки СТ.
Сучасний рівень наукових знань дозволяє вважати, що виявлені різноманітні порушення метаболізму СТ при СМ можуть бути зв'язані з безліччю молекулярних дефектів.
В останні роки проведене дослідження, що свідчить про знижену здатність до репарації ушкоджень ДНК, індукованих мутагенами, у клітках пробандів зі СМ, а також про порушення енергетичного обміну при СМ.
Вивчення патогенетичних механізмів порушень росту при СМ дозволило установити залучення в патологічний процес органів ендокринної системи і, у першу чергу, соматотропної функції гіпофіза і інсулярного апарата підшлункової залози. Виявлено показники соматотропину в 5-10 разів вище норми і пародоксальна реакція гормону на стандартний глюкозотолерантний тест (СГТТ). У дітей з легким плином захворювання відзначалося фізіологічне придушення активності соматотропного гормону на СГТТ. Патологічні типи цукрових кривих (предіабетичне і гіперактивні чи плоскі) і високий рівень інсуліну (перевищуючий норму в 5-10 кратному розмірі) також зареєстровані в хворих з повним, важкої симптомокомплексом хвороби. Існує припущення, що ендокринні розлади при СМ не тільки збільшують вагу захворювання, але в ряді випадків є одним з основних патогенетичних механізмів основних симптомів хвороби. Клінічна картина захворювання характеризується поразкою опорно-рухового апарата, серцево-судинної системи, око, органів подиху, центральної нервової системи (Рис 5).
Серед хворих зі СМ характерні дисгармоничність фізичного розвитку дітей, астенічна статура, високий ріст, довгі тонкі кінцівки, арахнодактілія кистей і стіп, гіпермобільність суглобів, уроджені контрактури кисті, воронкоподібна чи килевидна деформація грудної клітки, кифосколіоз, вузька грудна клітка, вальгусна деформація передпліч і гомілок, розгорнуті крила подвздошних кіст, чи плоскостопість плосковальгусна деформація стіп, вузький лицьовий кістяк з “готичним небом”, “пташине обличчя” із клювовидним носом, мікрогнатія, прогнатія, мікрогенія.
Пролапс митрального клапана, аневризму аорти з проградієнтним плином, порушення в провідній системі серця (порушення проведення порушення по правій ніжці пучка Гіса), аневризми судин печінки, головного мозку, артеріальна гіпертензія відносяться до основних судинних поразок при СМ.
У тісному зв'язку зі змінами серця і судин знаходиться патологія бронхіальної системи. Причиною для її розвитку є наявність у хворих зі СМ механічних факторів стискування дихальних систем при деформації грудної клітки, а також імовірність залучення в патологічний процес з’єднувально-тканинних структур легеневої тканини. Порушення органів зустрічається у виді спонтанного пневмотораксу, легеневої емфіземи, інфаркту легені, уродженого недорозвинення однієї з часток легені, полікистозу легень, уродженої буллезної емфіземи, двосторонніх бронхоектазів, порушення механіки подиху, здуття легеневої тканини, нерівномірності розподілу в легенях вдихуваного повітря.
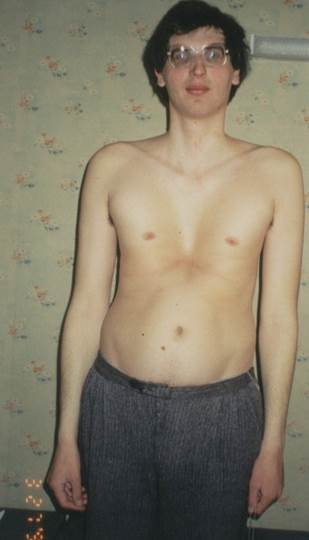

Рисунок 5. Синдром Марфана
Типовою патологією очей при СМ варто вважати вивих і подвивих кришталика внаслідок слабості циннової зв'язки, чи міопію гіперметропію високого ступеня, гетерохромія радужки, вторинна глаукома, розширення вен очного дна, чи дегенерація отслойка сітківки, уплощення роговиці, пресенильна катаракта, іридодонез, сферофакія, мікрофакія, мегалокорнєа.
У відношенні кістякових деформацій застосовується частіше консервативна терапія. Хірургічне втручання в зв'язку з деформацією грудини і кифосколіозом виправдано при погрозі здавлювання органів грудної клітки з розвитком хронічної дихальної чи серцевої недостатності.
Швидкий ріст хворих зі СМ може приводити до рецидиву кістякових деформацій, і це треба враховувати при виборі часу і характеру хірургічного втручання, пам'ятаючи, що СМ має прогредієнтний плин як усяке спадкове порушення обміну речовин.
СИНДРОМ ЕЛЕРСА-ДАНЛОСА
До складу з’єднувально-тканинних дисплазій входить велика група нозологічних варіантів синдрому Елерса-Данлоса (СЭД). СЭД – група спадкоємних захворювань з переважною поразкою шкіри і суглобів, в основі яких лежить дефект синтезу коллагену, вперше описаний у 1901 році дерматологом Ehlers, а потім Danlos у 1908 році. Його частота складає 1:100000 немовлят, однак стерті форми зустрічаються значно частіше.
В даний час виділяють більш десяти типів синдрому, що підкреслює його генетичну гетерогенність і клінічний поліморфізм.
Клінічні ознаки різних типів СЭД надзвичайно подібні між собою і практично немає критеріїв для їх клінічної диференцировки (табл.2). Вони з'являються гіперрозтяжністю та ранимістю шкіри, гіперрухливістю суглобів і симптомами гемморагічного діатезу. При цьому часто виявляється патологія опорно-рухового апарата, око, серцево-судинної і бронхо-легеневої систем, шлунково-кишкового тракту.
Для більшості типів СЭД характерна дуже м'яка, тендітна шкіра. Шкіра бархатиста, приємна на дотик, іноді тістоподібна, нерідко крізь неї просвічують судини, легкорастягується. На долонях і підошвах виражені зморшки.
Її можна підняти на 1,5-2 см у тих місцях, де це практично неможливо в здорової людини – на чолі, кінчику носа, вушних раковинах. При мінімальному травмуванні шкіри легко виникають “рвані” рані, що гояться вкрай повільно з утворенням атрофічних фляків з тонкою лискучою зморшкуватою поверхнею (“цигаркові” фляки). Часто формуються також келлоїдні фляки (звичайно післяопераційні); при цьому хірурги часто звертають на увагу на розбіжність швів. Під час операцій таку шкіру важко зіставити при зашиванні рані. Іноді відзначається наявність “псевдопухлин”, що утворяться в місцях найбільшого тиску (наприклад, в області колінних і ліктьових суглобів). Можливе утворення на передній поверхні гомілок вузликів, що формуються в результаті аномального росту жирової тканини і покритих фіброзними стінками, що містять кальцій. На шкірі в області гомілковостопних суглобів і гомілок хворих СЭД можливе утворення трофічних виразок.
Слизуваті оболонки при СЭД характеризуються аналогічними зі шкірою властивостями. Гіпереластичністю слизуватих оболонок порозумівається наявність у ряду пробандів симптому Горлина (здатність дістати мовою кінчик власного носа), що зустрічається в половини хворих із СЭД і в 10 % здорових облич. Результатами травм у хворих із СЭД можуть бути великі гематоми. Це зв'язано з поганою здатністю судин скорочуватися, у результаті чого вони довгостроково зяють: укол з діаметром голки довго залишається відкритим, розширюючи згодом.
Таблиця 2.

