




Посібник складено згідно з учбовою програмою дисципліни “Патологічна анатомія” для студентів лікувального, педіатричного та стоматологічного факультетів, лікарів – ітнернів, клінічних ординаторів, аспірантів, магістрів.
Розгянуті наступні теми:
Атеросклероз
Гіпертонічна хвороба та ішемічна хвороба серця
Ревматичні хвороби
Хвороби нервової системи
Хвороби органів дихання
Хвороби шлунково-кишкового тракту
Хвороби печінки
Захворювання нирок
Хвороби статевих органів та вагітності
Захворювання вагітності та післяпологового періоду
Патологія плода та новонароджених
Ендокринні хвороби
Згідно прогами «Патоморфологія» Київ, 2006р.
АТЕРОСКЛЕРОЗ
Атеросклероз (від грец. athere — каша та sklerosis — ущільнення) —це хронічнезахворювання, яке виникає в результаті порушення жирового та білкового обміну, характеризується ураженням артерій еластичного і еластично-м’язовогоо типу у вигляді вогнищевого відкладення в інтимі ліпідів та білків і реактивного розростання сполучної тканини.
Актуальність проблеми
Термін «атеросклероз» був запропонований Маршаном у 1904 р. Для дефініції захворювання, при якому склероз артерій зумовлений порушенням метаболізму ліпідів та білків, так званий «метаболічний артеріосклероз». Атеросклероз є різновидом артеріосклерозу. Термін «артеріосклероз» застосовують для позначення ущільнення і потовщення стінки артерій незалежно від причини і механізму його розвитку в результаті розростання в стінці судини сполучної тканини або утворення в ній патологічного білка гіаліна.
Види артеріосклерозу:
1. Віковий артеріосклероз – з віком в стінці артерій розростається сполучна тканина, артерії стають довшими на 14 % і приймають звивистий характер.
2. Акомодаційний (інволюційний) – при падінні артеріального тиску(наприклад – боталового протоку, артерії культі ампутованої кінцівки).
3. Гіпертонічний артеріосклероз розвиваються в результаті плазматичного просочування артеріол і утворення білка гіаліна.
4. Загальний артеріолосклероз. Наприкладв аорті при сифілітичному мезаотиті, висипному тифі, при введенні адреналіну, антибіотиків.
5. Алергічний – спостерігається при відкладанні імунних комплексів в стінку судин, або власне тканина стає алергеном.
6. Після петрифікації(Менкеберга)- кальцифікація інтими середнього калібру артерій майже виключно кінцівок(дуже рідко скроневої артерії, матки і яєчників).
7. Атеросклероз – порушення жиро-білкового обміну, зокрема холестерину з відкладанням його в стінку артерій з наступним фіброзом.
Частота атеросклерозу в усіх країнах світу за останні 50 років значно зросла і продовжує збільшуватися у всіх Європейських країнах. Тенденція до зниження захворювання за останнє десятиріччя відзначається лише в США. Захворювання в основному виявляється у другій половині життя. Ускладнення атеросклерозу є однією з найчастіших причин інвалідності і смертності у більшості країн світу. Хворі з проявами атеросклерозу знаходяться у стаціонарах практично будь-якого медичного профілю. Значне зниження смертельних ускладнень на американському континенті - це результат спільних зусиль не тільки кардіологів, фармакотерапевтів, але й епідеміологів. Таким чином, відомості про цю патологію необхідні також лікарям і медично-профілактичного профілю медицини. Знання морфологічного субстрату хвороби, особливо ранніх проявів атеросклерозу, дозволить фахівцю проводити не тільки грамотне патогенетично обгрунтоване лікування, але й визначити характер профілактичних заходів. Для кращого засвоєння даного розділу патології необхідні знання загальнопатологічних процесів, таких як: всі види альтерацій, порушення крово- і лімфообігу, компенсаторно-пристосувальних процесів.
Етіологія. Суперечка про природу атеросклеротичної бляшки серед фахівців різного профілю не затихає вже протягом цілого сторіччя. Запропоновано безліч гіпотез і теорій про причину розвитку атеросклерозу. Однак загальноприйнятої теорії виникнення атеросклерозу на сьогоднішній день немає. Однією з важливих ознак хвороби є широка варіабельність її проявів з точки зору гостроти і широти процесу, його поширеності за локалізацією у різних індивідуумів навіть в одній популяційній групі. Безліч чинників розглядаються як найважливіші фактори ризику розвитку атеросклерозу. Разом з тим, зустрічаються хворі з різко вираженими ознаками атеросклерозу, в яких неможливо виявити очевидні причини, які сприяють його розвитку, і що б можна було віднести до чинників ризику.
Захворюваність на атеросклероз підвищується з віком. У жінок нехарактерний його розвиток до клімактеричного періоду. Гіпертонія, підвищений рівень LDL-холестеролу(холестерин у ЛПНЩ) і цукровий діабет відносяться до особливих чинників ризику, незалежно від статі. В молодому віці розглядається як важливий чинник ризику паління цигарок. Менш важливими чинниками є повнота, сидячий спосіб життя, невисокий соціально-економічний статус.
Теорії патогенезу
1. Ліпопротеїдна теорія.
2. Імунологічна теорія
3. Вірусна теорія
4. Теорія реакції на пошкодження
5. Тромбогенна теорія.
6. Нейрон-метаболічна теорія.
Патогенез.
Основним фактором у патогенезі атеросклерозу є співвідношення ліпопротеїдів низької і дуже низької щільності(атерогенні ліпопротеїди) та ліпопротеїдів високої щільності (антиатерогенні ліпопротеїди), яке в нормі складає 4: 1 і значно зростає при атеросклерозі.
Епідеміологічними дослідженнями показано:
- у 2/3 випадків - атеросклероз обумовлений порушенням обміну ліпопротеїдів низкої щільності та ліпопротеїдів дуже низької щільності;
- у 1/3 випадків – виникнення захворювання пов'язують зі зниженням ліпопротеїдів високої щільності.
Незважаючи на наявність у клініці безлічі відеозображуючих технічних засобів, дуже тяжко простежити в динаміці прогресування атеросклерозу в однієї і тієї ж людини. Тому, майже вся інформація про розвиток атероматозних бляшок підпадає перевірці на тваринах (як спонтанний атеросклероз, так і атеросклероз, який розвивається в результаті застосування дієти з великою кількістю жиру).
Електронно-мікроскопічно встановлено, що в місцях, які схильні до розвитку атеросклерозу, на ранніх етапах його виявлення, між ендотеліальними клітинами верифікуються мігруючі в просвіт судини і з неї макрофаги. Накопичування фагоцитуючих макрофагів – це одна з ранніх морфологічних ознак хвороби. Молекулярні механізми прилипання макрофагів до ендотелію подібні до тих, які зустрічаються при гострому запаленні, але вони повністю ще не вивчені. Ендотеліальні клітини в ділянках формування атероматозної бляшки мають високу експресію адгезивних молекул, включаючи ICAM-1 та E-селектин. Можливо це один з ранніх молекулярних механізмів формування бляшки. Більшість прогресуючих атероматозних бляшок включають інфільтрати, які складаються з макрофагів, лімфоцитів і гладком’язових клітин і оточені здебільшого фіброзною тканиною. «Чинники росту», зокрема чинник росту, який виділяється з тромбоцитів (PDGF –platelet derived growth factor), стимулює проліферацію гладком’язових клітин інтими (міоінтимальні клітини) і продукцію в подальшому ними колагену, еластину та мукополісахаридів. PDGF секретується більшістю клітин сполучнотканинного походження, макрофагальної та ендотеліальної природи. Експериментально в культурі тканин показано, що PDGF прискорює ріст гладком’язових клітин і фібробластів, індукує подвоєння ДНК і, таким чином, сприяє прискоренню ділення клітин. Адгезивні молекули сприяють агрегації тромбоцитів, що супроводжується пошкодженням ендотеліальних клітин. Гемодинамічний тиск, особливо в місцях розгалуження судин сприяє прилипанню тромбоцитів і пошкодженню ендотелію. За певних умов, проміжок між ендотеліальними клітинами виявляється розширеним, і тоді з’являються або невеликі, або досить значні ділянки, позбавлені ендотеліальних клітин. Наступне вивільнення чинників росту, таких як PDGF, сприяє подальшій стимуляції проліферації і активації секреції гладком’язових клітин інтими. Наведені вище взаємовідносини між макрофагами, тромбоцитами, судинним ендотелієм сьогодні інтенсивно вивчаються багатьма фахівцями.
Ще Рудольф Вірхов підкреслював, що ліпіди – це важлива складова атероматозних пошкоджень. І зараз доведено, що підвищення рівня певних типів ліпопротеїнів істотно збільшує ризик розвитку атеросклерозу в різних людей.
Доведено, що підвищення в крові ліпопротеїнів з низькою питомою вагою, зокрема, LDL-холестеролу, є найважливішою і загальною причиною розвитку атероматозної бляшки. Рівень холестеролу регулюється як генетичними, так і екологічними чинниками. Ступінь смертності від атеросклеротичного пошкодження коронарних судин серця тісно пов’язаний з рівнем LDL-холестеролу. Підвищений ризик захворювань судин серця в Англії та інших північноєвропейських країнах зв’язують з великим вмістом жиру в харчовому раціоні мешканців цих країн. В країнах Середземномор’я, де менша пропорція насиченого жиру забезпечує енергію, смертність від захворювання коронарних судин низька. Разом з тим, встановлено, що харчовий розхід холестеролу порівняно мало впливає на рівень його в плазмі. Найцікавіші відомості про важливість LDL-холестеролу одержані при вивченні людини і тварин, в яких повністю або частково відсутні клітинні мембрани холестеролових рецепторів. Багато клітин мають рецептори, які розпізнають апопротеїнову частину LDL-молекули. Молекулярна структура LDL-рецептора визначена. Механізм, який контролює її синтез і переміщення на клітинну мембранну поверхню, достатньо вивчений. Більшість різних молекулярних аномалій успадковується як аутосомна домінантна ознака. Виявлено, що насиченість LDL-холестеролом особливо підвищена (понад 8 ммоль/л) в гетерозиготних хворих, особливо у тих, яким 40-50 років і які мають захворювання коронарних судин. Гомозиготні хворі, які зустрічаються дуже рідко (приблизно 1 на 1 млн мешканців), з дефіцитом рецепторів, в основному помирають в дитячому і підлітковому віці від атеросклеротичних уражень коронарних судин серця. Точний механізм, шляхом якого підвищений рівень LDL-холестеролу прискорює розвиток атеросклерозу, ще не визначений. Високий рівень холестеролу, який циркулює в крові, може підвищити вміст холестеролу в мембранах ендотелію. Підвищення його в мембранних структурах веде до зниження їхньої пружності і сприяє пошкодженню. На сьогодні доведено, що коли LDL-холестерол окислюється макрофагами, адгезованими на ендотелію судини, вільні радикали можуть пошкоджувати підлеглі гладком’язові клітини. Крім того, хронічна гіперхолестеролемія сприяє підвищенню секреції ендотелієм у величезній кількості чинників росту, таких як PDGF.
Інтерес представляють також дослідження з обміном високомолекулярного ліпопротеїду HDL-холестеролу (холестерин ЛПВЩ). HDL-холестерол втягується в холестероловий транспорт, прямуючи з периферійних тканин в печінку. В літературі наведено декілька вірогідних епідеміологічних досліджень, в яких показано, що високий вміст HDL-холестеролу в клітинах печінки пов’язаний з пониженням ризику розвитку атеросклеротичних змін коронарних судин серця. Дослідження в цьому напрямку вважаються перспективними.
Незважаючи на те, що вміст тригліцеридів у крові відноситься до слабких чинників ризику розвитку атеросклерозу, необхідно враховувати його, оскільки спадкові аномалії ліпідного метаболізму пов’язані з підвищеним рівнем холестеролу і тригліцеридів.
Інші патогенетичні чинники розвитку атеросклерозу. Гістологічні дослідження атероматозних змін у людини і тварини показали, що фібрин і тромбоцити відносяться до важливих складових ранніх пошкоджень. На сьогодні існують докази, що підвищений ризик ІХС пов’язаний з підвищенням рівня фактора зсідання VII. Ранні зміни тромботичної формації включають активацію тромбоцитів з наступною адгезією до субендотеліального колагену. Агенти, які стимулюють активацію тромбоцитів, колаген, тромбін, тромбоксан А2, аденозин фосфат, норадреналін, тобто агенти-вазопресори. Зараз відомо, що ці фактори стимулюють глікопротеїнові рецептори на мембранах тромбоцитів. Повна назва цих рецепторів – тромбоцитарний глікопротеїн IIВ/IIIА. Малі дози аспірину, які призначаються хворим з клінічними проявами атеросклеротичного ураження коронарних судин і мають напевно цілющий ефект, інгібують дію тромбоксану А2. Сьогодні тривають пошуки інших засобів інгібіції рецепторів глікопротеїну IIВ/IIIА.
Коротко фактори ризику атеросклерозу можна схематично відобразити так:
 | ||||
 | ||||
| ||||
|



 АА
АА

| ||
| ||
|
Патологічна анатомія і морфогенез
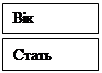
При атеросклерозі в інтимі аорти і артерій появляються кашоподібний жиробілковий детрит (аthеге) і осередкове розростання сполучної тканини (sclerosis), що призводить до формування атеросклеротичної бляшки (див. малюнок), яка звужує просвіт судини. Уражаються артерії еластичного і м’язово-еластичного типу, тобто артерії великого і середнього калібру, значно рідше в процес втягуються дрібні артерії м’язового типу.

Атеросклеротична бляшка в поперечному розрізі
Розрізняють наступні стадії морфогенезу атеросклерозу:
— доліпідна;
— ліпоїдоз;
— ліпосклероз;
— атероматоз;
— виразкування;
— атерокальциноз.
Доліпідна стадія макроскопічно не визначається. Мікроскопічно спостерігається:
— вогнищеве ураження (аж до повної деструкції) ендотелію і підвищення проникності мембран інтими, що веде до накопичування у внутрішній оболонці білків плазми, фібриногену (фібрину) і утворення плоских пристінкових тромбів;
— накопичення кислих глікозаміногліканів в інтимі, мукоїдне набухання внутрішньої оболонки, поява в ній ліпопротеїдів дуже низької і низької щільності, холестерину, білків;
— руйнування еластичних і колагенових волокон, проліферація гладком’язових клітин.
Для виявлення цієї стадії необхідне застосування тіазинових барвників. Наприклад, завдяки застосуванню забарвлення препарату толуїдиновим синім (тіоніном), можна спостерігати появу пурпурного забарвлення (явище метахромазії) в ділянках ранньої дезорганізації сполучної тканини.
Стадія ліпоїдозу характеризується вогнищевою інфільтрацією інтими ліпідами (холестерином), ліпопротеїдами, що веде до утворення жирових (ліпідних) плям і смуг. Макроскопічно такі жирові плями у вигляді ділянок жовтого кольору, інколи можуть зливатися і утворювати плоскі подовжені смуги, які не підносяться над поверхнею інтими. В цих ділянках при застосуванні барвників на жир, наприклад, судан III, IV, жировий червоний О та інші, в надлишку виявляються ліпіди. Ліпіди накопичуються в гладко-м’язових клітинах і макрофагах, які отримали назву пінявих, або ксантомних, клітин (від грец. xаnthos — жовтий). В ендотелії також появляються ліпідні включення, що свідчить про інфільтрацію інтими ліпідами плазми крові. Спостерігається набухання і руйнування еластичних мембран. Перш за все жирові плями і смужки появляються в аорті і в місцях відходження її гілок, після цього - у великих артеріях. Поява подібних плям ще не означає наявність атеросклерозу, оскільки появу ліпідних плям можна спостерігати в ранньому дитячому віці не тільки в аорті, але й у вінцевих артеріях серця. З віком ліпідні плями, так звані прояви «фізіологічного раннього ліпідозу», в більшості випадків зникають і не є джерелом розвитку подальших атеросклеротичних змін. Аналогічні зміни в судинах у молодих людей можна виявити при деяких інфекційних захворюваннях.

Тонка фіброзна покришка атеросклеротичної бляшки між стрілками відділяє ліпідне м'яке ядро від просвіту судини.
При ліпосклерозі відбувається проліферація фібробластів, ріст яких стимулює руйнування макрофагів (ксантомних клітин) і розростання в інтимі молодої сполучної тканини. Наступне дозрівання цієї тканини супроводжується формуванням фіброзної бляшки. Макроскопічно фіброзні бляшки являють собою щільні, круглої або овальної форми утворення білого або жовтувато-білого кольору, які підносяться над поверхнею інтими. Застосуванням барвників на жир у фіброзних бляшках виявляються ліпіди. Ці бляшки звужують просвіт, що супроводжується порушенням надходження крові (ішемії) до органа або його частини. Найчастіше фіброзні бляшки спостерігаються в черевній аорті, в гілках, які відходять від аорти, в артеріях серця, мозку, нирок, нижніх кінцівок, сонних артеріях, тощо.
Ліпідні маси атеросклеротичної бляшки повністю
заповнюють просвіт судини.
При атероматозі ліпідні маси, які розташовані в центральній частині бляшки, і прилеглі колагенові та еластичні волокна розпадаються. В утвореній дрібнозернистій аморфній масі (вказані на малюнку товстою стрілкою) виявляються кристали холестерину і жирних кислот, уламки еластичних і колагенових волокон, крапельки нейтральних жирів (атероматозний детрит). Виявляється багато ксантомних клітин, лімфоцитів і плазмоцитів. Атероматозні маси відокремлені від просвіту судини шаром зрілої, гіалінізованої сполучної тканини (покришка бляшки).
Прогресування атероматозних змін веде до деструкції покришки бляшки. Цей період характеризується великою кількістю різних ускладнень. Наступає стадія виразки, яка супроводжується утворенням атероматозної виразки. Краї такої виразки підриті, нерівні, дно утворене м’язовим, а інколи - адвентиційним шаром стінки судини. Дефект інтими нерідко покривається тромботичними нашаруваннями. В результаті некрозу глибоких шарів стінки судини може формуватися аневризма (випинання стінки). Нерідко кров відшаровує інтиму від середнього шару, і тоді виникають розшаровуючі аневризми. Небезпека цих ускладнень полягає в можливості розриву або аневризми, або ж стінки судини в місцях виникнення атероматозних виразок. Атероматозні маси можуть вимиватися течією крові і формувати емболи.
Атерокальциноз характеризується відкладенням у фіброзних бляшках солей кальцію, тобто їх звапнінням (петрифікацією). Це завершальна стадія атеросклерозу. Разом з тим, необхідно пам’ятати, що відкладання солей кальцію може спостерігатися і на його більш ранніх стадіях. Бляшки набувають кам’янистої щільності, стінка судини в місці петрифікації різко деформується. Солі кальцію відкладаються в атероматозні маси, у фіброзну тканину, в проміжну речовину між еластичними волокнами.
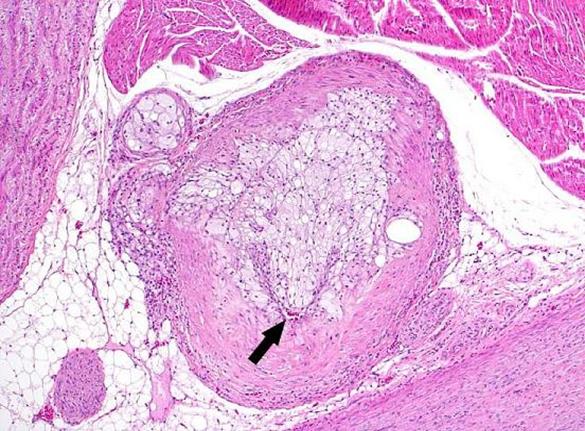
Патоморфоз змін в атеросклеротичній бляшці:
А - ступінь стенозу 90%.
В, С - невеликі канали реканалізації в капілярах.
D – запальна інфільтрація в адвентиції судини.
Е – розростання капілярів в адвентиції судини.
Клінічний перебіг. Атеросклероз – це хронічне рецидивуюче захворювання. Для нього характерний хвилеподібний перебіг, який включає в собі чергування трьох основних фаз:
— прогресування;
— стабілізацію;
— регресування процесу.
Хвилеподібність перебігу полягає в нашаруванні ліпідозу на старі зміни – ліпосклерозу, атероматозу і атерокальцинозу. Більш виражені зміни відмічаються в місцях первинного ушкодження (черевний відділ аорти, дуга аорти). При регресуванні процесу можливе часткове розсмоктування ліпідів за допомогою макрофагів.
Ускладнення атеросклерозу. Незалежно від локалізації атеросклеротичних змін розрізняють дві групи ускладнень: хронічні та гострі.
Хронічні ускладнення. Атеросклеротична бляшка, випинаючись у просвіт судини, веде до звуження (стенозу) його просвіту (стенозуючий атеросклероз). Оскільки формування бляшки в судинах процес повільний, виникає хронічна ішемія в зоні кровопостачання даної судини. Хронічна судинна недостатність супроводжується гіпоксією, дистрофічними і атрофічними змінами в органі і розростанням сполучної тканини. Повільна оклюзія судин призводить в органах до дрібновогнищевого склерозу.
Гострі ускладнення. Вони зумовлені виникненням тромбів, емболів, спазмом судин. Виникає гостра оклюзія судин, яка супроводжується гострою судинною недостатністю (гостра ішемія), що призводить до розвитку інфарктів органів (наприклад, інфаркт міокарда, сіре розм’якшення мозку, гангрена кінцівки тощо). Інколи може спостерігатися розрив аневризми судини зі смертельним виходом внаслідок крововиливу (наприклад, у головний мозок з наступним набряком його та дислокацією) чи масивної кровотечі з розвитком гострої постгеморагічної анемії при враженні крупних судин (наприклад, аневризма аорти).
КЛІНІКО-МОРФОЛОГІЧНІ ФОРМИ АТЕРОСКЛЕРОЗУ
Залежно від переважаючої локалізації атеросклеротичних змін в судинах, ускладнень і виходу, до яких ці зміни ведуть, виділяють наступні клініко-анатомічні форми атеросклерозу:
— атеросклероз аорти;
— атеросклероз вінцевих артерій серця (ішемічна хвороба серця);
— атеросклероз артерій головного мозку (цереброваскулярні захворювання);
— атеросклероз артерій нирок (ниркова форма);
— атеросклероз артерій кишки (кишкова форма);
— атеросклероз артерій нижніх кінцівок.

1- стрілками вказані атеросклеротичні бляшки в аорті, у вставці мікроскопічна картина атероматозної бляшки;
2- ліпоматоз підшлункової залози при ожирінні (фактор ризику атеросклерозу);
3- кістознозмінені нирки;
4 - атеросклеротично-зморщена нирка (великогорбиста).
АТЕРОСКЛЕРОЗ АОРТИ
Атеросклероз аорти – це найчастіша форма атеросклерозу. Найбільш різко атеросклеротичні зміни виражені в черевному відділі і характеризуються в основному атероматозом, виразкуванням, атерокальцинозом. В результаті тромбозу, тромбоемболії та емболії атероматозними масами при атеросклерозі аорти часто спостерігаються інфаркти (наприклад, нирок) і гангрени (наприклад, кишки, нижньої кінцівки). 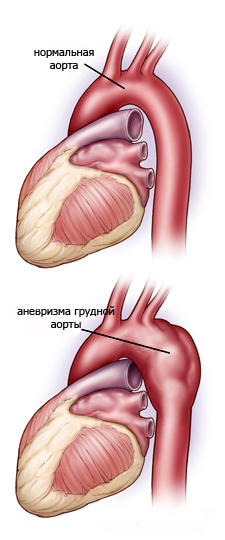
При атеросклерозі в аорті часто розвиваються аневризми. Розрізняють циліндричну, мішкоподібну аневризми аорти.
 При відсепаровці інтими аорти і спрямовування течії крові між шарами стінки аорти формується розшаровуюча аневризма аорти(a. dissecans). Утворення аневризми небезпечне її розривом і крововиливами. Тривало існуюча аневризма аорти призводить до атрофії навколишніх тканин –узур (наприклад, груднини, тіл хребців).
При відсепаровці інтими аорти і спрямовування течії крові між шарами стінки аорти формується розшаровуюча аневризма аорти(a. dissecans). Утворення аневризми небезпечне її розривом і крововиливами. Тривало існуюча аневризма аорти призводить до атрофії навколишніх тканин –узур (наприклад, груднини, тіл хребців).
Атеросклероз вінцевих артерій серця лежить в основі ішемічної його хвороби (див. «Ішемічна хвороба серця»).
Атеросклероз артерій головного мозку є основою цереброваскулярних захворювань (див. “Цереброваскулярні захворювання”). Тривала ішемія головного мозку на грунті стенозуючого атеросклерозу мозкових артерій призводить до дистрофії і атрофії кори мозку, розвитку атеросклеротичного недоумства.
При атеросклерозі ниркових артерій звуження просвіту бляшкою здебільшого спостерігається в місці розгалуження основного стовбура або поділу його на гілки першого і другого порядку. Частіше процес односторонній, рідше — двосторонній. В нирках розвиваються або клиноподібні ділянки атрофії паренхіми з колапсом строми і заміщенням цих ділянок сполучною тканиною, або інфаркти з наступною їх організацією і формуванням втягнутих рубців або кіст. Виникає крупногорбиста атеросклеротична зморщена нирка (атеросклеротичний нефросклероз), функція якої страждає мало, бо більша частина паренхіми залишається збереженою. В результаті ішемії ниркової тканини при стенозуючому атеросклерозі ниркових артерій у ряді випадків розвивається симптоматична (вазо-ренальна) гіпертонія.
Атеросклероз артерій кишки, ускладнений тромбозом, веде до гангрени кишки з наступним розвитком перитоніту. Частіше страждає верхня брижова артерія.
При атеросклерозі артерій кінцівок частіше уражаються стегнові артерії. Процес тривалий часом перебігає безсимптомно завдяки розвитку колатералей. Однак, при наростаючій недостатності колатералей розвиваються атрофічні зміни м’язів, охолодження кінцівки, з’являються характерні болі при ходьбі — переміжне кульгання. Якщо атеросклероз ускладнюється тромбозом, розвивається гангрена кінцівки — атеросклеротична гангрена.

Розшаровуюча аневризма аорти.
ГІПЕРТОНІЧНА ХВОРОБА
Гіпертонічна хвороба (синоніми: первинна, або есенціальна, ідіопатична гіпертензія) –хронічне захворювання, основною клінічною ознакою якого є тривале і стійке підвищення артеріального тиску (гіпертензія).
Гіпертонія за етіологією може бути класифікована відповідно:
— «первинна» (ідіопатична) – причина невідома;
— «вторинна» або симптоматична гіпертензія, яка є проявом багатьох захворювань нервової, ендокринної систем, патології нирок і судин.
СИМПТОМАТИЧНI ГІПЕРТЕНЗІЇ

Гіпертензія (гіпертонія) – найчастіша причина високої захворюваності і смертності у всьому світі. Більшість випадків гіпертонії класифікується як “первинна”, але необхідно пам’ятати про можливість невиявлення причини внаслідок недостатнього обстеження хворого. Прийнято вважати, що гіпертонічна хвороба, як і атеросклероз, є хворобою урбанізації і широко поширена в економічно розвинених країнах, які зазнають зростаючого напруження психоемоціональної сфери. Гіпертонічну хворобу називають “хворобою невідреагованих емоцій”. При епідеміологічному дослідженні Африканського континенту, а також в деяких районах, розташованих в східній частині Тихого океану, серед мешканців відзначено незвичайно низький середній АТ. Однак, в Східній і Північній Африці зареєстрований високий рівень захворюваності на гіпертонічну хворобу з тенденцією до прогресування. Епідеміологічні дані свідчать про позитивну кореляційну залежність між вагою і, як систолічним, так і діастолічним АТ. Цей зв’язок особливо сильний у молодих людей, але зменшується в літніх. Відзначено, що у гіпертоніків, які втрачають масу, знижується АТ. Висловлюється гіпотеза про те, що високий АТ передається спадково, однак точних даних не приводиться. АТ хворих і їхніх безпосередніх дітей знаходиться в залежності, в той час як у батьків і прийомних дітей такої залежності не спостерігається. Кореляція АТ у гомозиготних близнюків висока, а в гетерозиготних — низька.
Допомогу хворим на гіпертонічну хворобу і вторинну гіпертензію надають фахівці різного профілю: терапевти, нефрологи, кардіологи, невропатологи, нейрохірурги та інші спеціалісти. Знання морфологічного субстрату первинної і вторинної гіпертензії необхідні фахівцям медичного і медико-профілактичного профілю. Для кращого засвоєння даної теми необхідні знання наступних загальнопатологічних процесів: всі види альтерацій, порушення крово - і лімфообігу, компенсаторно-пристосувальні процеси.
Патогенез. Загальноприйнятої теорії походження і розвитку гіпертонічної хвороби на сьогодні немає. Ключова ознака тривалої первинної гіпертензії – це підвищення периферичної судинної резистентності. Численні ретельні клінічні і фізіологічні дослідження вказують на те, що існує безліч механізмів, які призведуть до розвитку первинної гіпертонії. З них сьогодні загальноприйнятими вважаються три основні патофізіологічні механізми, які включають:
— натрієвий гомеостаз;
— симпатичну нервову систему;
— ренін-ангіотензин-альдостеронову систему.
Натрієвий гомеостаз. Відзначено, що першими змінами, які виявляються, є уповільнена ниркова екскреція натрію. Натрієва затримка супроводжується збільшенням об’єму і швидкості кровотоку, що зумовлено збільшенням серцевого викиду. Периферична ауторегуляція підвищує судинну резистентність і в підсумку зумовлює гіпертонію. У хворих з первинною гіпертонією Nа+-К+-транспорт змінений у всіх клітинах крові. Крім того, плазма крові гіпертоніків при її переливанні може пошкоджувати Nа+-К+-транспорт в клітинах крові здорових людей. Це говорить про наявність у хворих (із зменшеною натрієвою екскрецією) циркулюючих в крові субстанцій, які інгібують Nа+-транспорт в нирках та інших органах. Загальний рівень Nа+в організмі позитивно корелює з АТ у гіпертоніків і не корелює у досліджених нормотоніків (контрольна група). В більшості здорових дорослих людей виявляються незначні зміни АТ, які залежать від вживання солі з їжею. Деякі гіпертоніки класифікуються, як «первинно-сольові», але природа змін, що лежать в основі гіпертонії у цих хворих, невідома. Відомо, що підвищений перехід Nа+в ендотеліальні клітини артеріальної стінки може також підвищувати і внутрішньоклітинний вміст Са2+. Це сприяє підвищенню судинного тонусу і звідси, отже, периферичного судинного опору.
Симпатична нервова система. Артеріальний тиск – це похідна загального периферичного судинного опору і серцевого викиду. Обидва ці показники знаходяться під контролем симпатичної нервової системи. Виявлено, що рівень катехоламінів у плазмі крові у хворих на первинну гіпертензію підвищений у порівнянні з контрольною групою. Рівень циркулюючих катехоламінів дуже варіабельний і може змінюватися з віком, надходженням Nа+до організму, у зв’язку зі станом і фізичним навантаженням. Крім того, встановлено, що у хворих на первинну гіпертонію спостерігається тенденція до більш високого вмісту норадреналіну в плазмі, ніж у молодих людей контрольної групи з нормальним АТ.
Ренін-ангіотензин-альдостеронова система. Ренін утворюється в юкстагломерулярному апараті нирок, дифундує у кров через “виносні артеріоли”. Ренін активує плазматичний глобулін (називається “реніновий субстрат”, або ангіотензин) для звільнення ангіотензину I. Ангіотензин I перетворюється в ангіотензин II під впливом ангіотензин-трансферази. Однак, тільки у невеликої кількості хворих на первинну гіпертонію має місце підвищений рівень реніну у плазмі крові, таким чином, немає простого прямого співвідношення між активністю плазматичного реніну і патогенезом гіпертонії. Є відомості, що ангіотензин може стимулювати симпатичну нервову систему центрально. Багато хворих піддаються лікуванню за допомогою інгібіторів ангіотензин-трансферази - таких як каптопріл, еналопріл, енал, котрі інгібують ферментативне перетворення ангіотензину I в ангіотензин II. Декілька терапевтичних експериментів виявили, що інгібітори ангіотензин-трансферази, введені незабаром після гострого інфаркту міокарда знижують смертність, як припускається, в результаті зменшення міокардіальної дилатації.
Нещодавно виявлені асоціації між мутацією генів, які кодують продукцію ангіотензину I, ангіотензин-трансферази і деяких рецепторів для ангіотензину II, і розвитком первинної гіпертонії. Встановлений також зв’язок між поліморфізмом гена, який кодує продукцію ангіотензин-трансферази та “ідіопатичною” серцевою гіпертрофією у хворих з нормальним артеріальним тиском. Разом з тим, точний механізм змін структури генів поки що невідомий.
Патологічна анатомія. Морфологічні прояви гіпертонічної хвороби залежать від характеру і тривалості її перебігу. За характером перебігу хвороба може перебігати злоякісно(злоякісна гіпертензія) і доброякісно(доброякісна гіпертензія).
При злоякісній гіпертензії домінують прояви гіпертонічної кризи, тобто різкого підвищення артеріального тиску у зв’язку зі спазмом артеріол. Морфологічні прояви гіпертонічної кризи:
— гофрованість і деструкція базальної мембрани ендотелію (розташування його у вигляді частоколу в результаті спазму артеріоли);
— плазматичне просякання або фібриноїдний некроз її стінки;
— тромбоз, сладж-феномен.
При цій формі часто розвиваються інфаркти, крововиливи, на сьогодні злоякісна гіпертонія зустрічається рідко, переважає доброякісно і поволі перебігаюча гіпертонічна хвороба.
При доброякісній формі гіпертонічної хвороби розрізняють три стадії, які мають певні морфологічні відмінності:
— доклінічну;
— виражених поширених морфологічних змін артеріол і артерій;
— вторинних змін внутрішніх органів, зумовлених змінами судин і порушенням внутрішньоорганного кровообігу.

Разом з тим, в будь-якій стадії доброякісної гіпертензії може виникнути гіпертонічна криза з характерними для неї морфологічними проявами.
Доклінічна стадія гіпертонічної хвороби характеризується періодичним і тимчасовим підвищенням артеріального тиску (транзиторна гіпертензія). При мікроскопічному дослідженні виявляють помірну гіпертрофію м’язового шару та еластичних структур артеріол і дрібних артерій,спазм артеріол. Клінічно і морфологічно виявляють помірнугіпертрофію лівого шлуночка серця.
Стадія виражених поширених морфологічних змін артеріол і артерій є результатом тривалого підвищення артеріального тиску. В цій стадії виникають морфологічні зміни в артеріолах, артеріях еластичного, м’язово-еластичного і м’язового типів, а також у серці.
Найхарактернішою ознакою гіпертонічної хвороби є зміни артеріол. В артеріолах виявляється плазматичне просякання, яке завершується артеріолосклерозом і гіалінозом.
Плазматичне просякання артеріол і дрібних артерій розвивається у зв’язку з гіпоксією, зумовленою спазмом судин, що веде за собою пошкодження ендотеліоцитів, базальної мембрани, м’язових клітин і волокнистих структур стінки. В подальшому білки плазми ущільнюються і перетворюються в гіалін. Розвивається гіаліноз артеріол або артеріолосклероз. Найчастіше плазматичного просякання і гіалінозу зазнають артеріоли і дрібні артерії нирок, головного мозку, підшлункової залози, кишки, сітківки ока, капсули наднирників.
В артеріях еластичного, м’язово-еластичного і м’язового типів виявляється еластоз і еластофіброз. Еластоз і еластофіброз — це послідовні стадії процесу і являють собою гіперплазію і розщеплення внутрішньої еластичної мембрани, що розвивається компенсаторно у відповідь на стійке підвищення артеріального тиску. В подальшому відбувається загибель еластичних волокон і заміщення цих локусів колагеновими волокнами, тобто склероз.


Склероз і потовщення стінки судини при гіпертонічній хворобі.
Стінка судин потовщується, просвіт звужується, що веде до розвитку хронічної ішемії в органах. В крупних судинах еластичного типу спостерігається атеросклероз. При цьому він швидко прогресує і вираженість його більша, ніж при чистому атеросклерозу. Зміни в артеріолах і артеріях м’язово-еластичного і м’язового типів створюють передумови для розвитку третьої стадії гіпертонічної хвороби. В цій стадії маса серця досягає 900-1000 г, а товщина стінки лівого шлуночка має 2-3 см. У зв’язку з порушенням трофіки міокарда (в умовах кисневого голодування) розвивається дифузний дрібновогнищевий кардіосклероз.
Остання стадія гіпертонічної хвороби, або стадія вторинних змін внутрішніх органів, зумовлена патологією судин і порушенням внутрішньоорганного кровообігу.
Ці вторинні зміни можуть виявлятися або дуже швидко в результаті спазму, тромбозу, фібриноїдного некрозу стінки судини і завершуються крововиливами або інфарктами, або ж можуть розвиватися поволі за рахунок гіалінозу і артеріолосклерозу і призводити до атрофії паренхіми і склерозу органів.
На підставі домінування судинних, геморагічних, некротичних і склеротичних змін в серці, мозку, нирках при гіпертонічній хворобі виділяють серцеву, мозкову і ниркову клініко-морфологічні її форми.
Серцева форма гіпертонічної хвороби разом із серцевою формою атеросклерозу складають сутність ішемічної хвороби серця (див. «Ішемічна хвороба серця»).
Мозкова форма гіпертонічної хвороби розглядається в розділі щодо цереброваскулярних захворювань.
Ниркова форма гіпертонічної хвороби характеризується як гострими, так і хронічними змінами.
До гострих змін відносяться інфаркти нирок і артеріолонекроз нирок, які пов’язані з тромбоемболією або тромбозом артерій. Артеріолонекроз нирок є морфологічним проявом злоякісної гіпертонії. Окрім артеріол, фібриноїдного некрозу зазнають капілярні петлі клубочків, в стромі виникають набряк і геморагії, в епітелії канальців — білкова дистрофія. У відповідь на некроз в артеріолах, клубочках і стромі розвивається клітинна реакція і склероз. Нирки виглядають дещо зменшеними в розмірах, строкатими, їх поверхня дрібногранулярна. Артеріолонекроз призводить до гострої ниркової недостатності і закінчується зазвичай смертельно.
Найхарактерніші зміни виявляються в нирках при доброякісному перебігові гіпертонічної хвороби. Ці зміни зумовлені недостатнім кровопостачанням, тобто хронічною ішемією. В результаті недостатнього кровопостачання і гіпоксії канальцева частина більшості нефронів атрофується і заміщається сполучною тканиною, яка розростається також навколо «мертвих» клубочків. На поверхні нирок з’являються множинні дрібні осередки западання. Нефрони у відносно збережених клубочках гіпертрофуються і виступають над нирковою поверхнею. Нирки різко зменшуються в розмірі (практично вдвічі), щільні, поверхня дрібнозерниста, паренхіма рівномірно стоншена, особливо кіркова речовина. Маса нирок може досягати 50-60 г. Такі нирки називаються первинно-зморщеними. «Первинно» — тому що зменшення нирок відбувається від нормальних розмірів, в той час як у всіх інших випадках (при запаленні, дистрофічних процесах) нирки спочатку збільшуються в обсязі, а після цього вторинно відбувається їхнє зменшення. Ще одна назва хвороби нирок “артеріоло-склеротичний нефросклероз” вказує, що в основі захворювання першочергово лежить ураження артеріол. Хворі найчастіше помирають при цій формі від хронічної ниркової недостатності (азотемічної уремії).
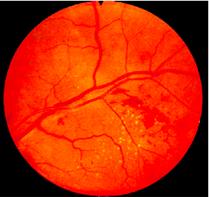 Зміни очей при гіпертонічній хворобі вторинні і пов’язані з типовими змінами судин. Ці зміни проявляються у вигляді набряку соска зорового нерву, крововиливів, відшарування сітківки, у важких випадках - її некрозом і важкими дистрофічними змінами нервових клітин гангліозного шару.
Зміни очей при гіпертонічній хворобі вторинні і пов’язані з типовими змінами судин. Ці зміни проявляються у вигляді набряку соска зорового нерву, крововиливів, відшарування сітківки, у важких випадках - її некрозом і важкими дистрофічними змінами нервових клітин гангліозного шару.
Гипертоническая ретинопатия: тромбозы вен и геморрагии сетчатки (по J.D. Swales)
Причини смерті. Найчастішими причинами смерті є серцева недостатність в результаті дифузного кардіосклерозу (в гострих випадках – інфаркт міокарда), хронічна ниркова недостатність (азотемічна уремія), крововилив у мозок.
ІШЕМІЧНА ХВОРОБА СЕРЦЯ
Ішемічна хвороба серця (ІХС) – група захворювань, зумовлених абсолютною або відносною недостатністю коронарного кровобігу. Тому ішемічна хвороба – це коронарна хвороба серця. Ішемічна хвороба на сьогодні поширена у всьому світі, особливо в економічно розвинених країнах. Небезпека ішемічної хвороби серця полягає в раптовій смерті. На її частку припадає приблизно 2/3 випадків смерті від серцево-судинних захворювань. Хворіють частіше чоловіки у віці 40-65 років.
Фактори ризику ішемічної хвороби серця:
- дисліпопротеїдемія;
- табакопаління;
- гіпертензія;
- ожиріння, гіподинамія;
- вік, стать;
- спадковість.
Ішемічна хвороба серця – це серцева форма атеросклерозу і гіпертонічної хвороби, яка проявляється морфологічно ішемічною дистрофією міокарда, інфарктом міокарда, кардіосклерозом.
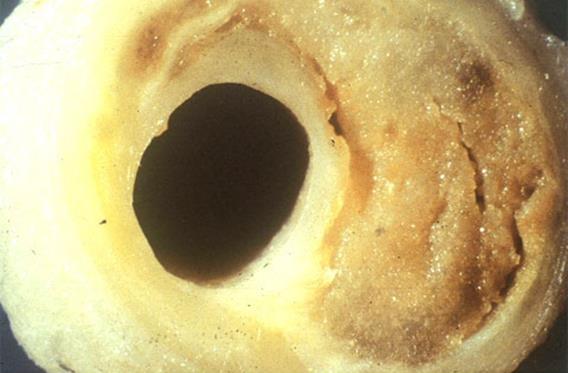
Атеросклероз коронарної судини (в правій половині судини масивна атеросклеротична бляшка з чітко виразними атероматозними масами)
Ішемічна хвороба серця перебігає хвилеподібно, супроводжуючись коронарними кризами, тобто епізодами гострої (абсолютної) коронарної недостатності, які виникають на тлі хронічної (відносної) недостатності коронарного кровообігу. У зв’язку з цим розрізняють гостру і хронічну форми ішемічної хвороби серця.
Класифікація ішемічної хвороби серця(схематично):
Гостра ІХС:
- раптова коронарна смерть
- гостра вогнищева іішемічна дистрофія міокарда
- інфаркт міокарду
Хронічна ІХС:
- постінфарктний крупновогнищевий кардіосклероз
- дифузний дрібновогнищевий кардіосклероз
Гостра ішемічна хвороба серця морфологічно проявляється ішемічною дистрофією міокарда та інфарктом міокарда, хронічна ішемічна хвороба серця (ХІХС) — кардіосклерозом (дифузним дрібновогнищевим і постінфарктним крупновогнищевим), який ускладнюється інколи хронічною аневризмою серця.
Ішемічна дистрофія міокарда, або гостра вогнищева дистрофія міокарда, (клінічно: стенокардія напруги чи спокою) розвивається при відносно короткочасних епізодах коронарної кризи, коли виникають характерні зміни електрокардіограми при відсутності некрозу міокарда (відсутнє підвищення активності трансаміназ, лактатдегідрогенази). Міокард в’ялий і блідий, в ділянках ішемії інколи строкатий і набряклий. Нерідко в коронарній артерії виявляється свіжий тромб.
Макроскопічно при обробці поверхні розрізу міокарда розчином солі тетразолію, теллурита калію, ділянки ішемії виглядають світлими на темному тлі незміненого міокарда.
Мікроскопічно знаходять дилатацію капілярів, стаз і сладж-феномен еритроцитів, набряк інтерстиційної тканини, периваскулярні крововиливи, скупчення лейкоцитів по периферії зони ішемії. М’язові волокна втрачають смугастість, позбавлені глікогену, вони інтенсивно забарвлюються еозином, фуксином, піроніном і реактивом Шиффа, що свідчить про некробіотичні зміни. Пофарбовані акридиновим оранжевим вони дадуть в люмінесцентному мікроскопі не оранжеве, а зелене свічення, що дозволяє відрізнити зону ішемії від інтактного міокарда. Поляризаційно-оптично виявляється безліч контрактур.
Ранні електронно-мікроскопічні та гістохімічні зміни зводяться до зменшення числа гранул глікогену, зниження активності окисно-відновних ферментів (особливо дегідрогеназ та діафораз), набухання і деструкції мітохондрій та саркоплазматичної сітки. Гістохімічно при використанні солей тетразолію чи теллурита калію в ділянках ішемії активність окисно-відновних ферментів різко послаблена, і тому зерна формазану, а також відновлений теллур не випадають.
Ці зміни, пов’язані з порушенням тканинного дихання, підсиленням анаеробного гліколізу і роз’єднанням дихання і окисного фосфорилювання, появляються вже через декілька хвилин після початку ішемії.
Наслідки гострої ішемії міокарду:

Ускладненням ішемічної дистрофії міокарда найчастіше є гостра серцева недостатність, вона і є безпосередньою причиною смерті.

