




’орошо известно, что даже в пределах одной и той же реакции относительна€ реакционна€ способность родственных функций может ощутимо зависеть от конкретных особенностей используемого реагента. ѕоэтому правильный вы≠бор реагента из уже имеющегос€ набора (или рациональный дизайн нового реагента) может оказатьс€ наиболее эффективным путем обеспечени€ требу≠емой селективности данного превращени€. ¬от характерный пример.
¬ конце 1940-х годов в практику органической химии был внедрен прин≠ципиально новый и мощный восстановитель Ч алюмогидрид лити€. ќтвле≠ка€сь от деталей механизма восстановлени€ функциональных групп с по≠мощью этого реагента, мы можем прин€ть, что суть реакции состоит в нук-леофильной атаке гидрид-иона Ќ- на субстрат. ѕон€тно, что субстратом по отношению к такому реагенту должен быть электрофил, и в принципе любые соединени€, содержащие электрофильные группы, должны поддаватьс€ вос≠становлению алюмогидридом лити€. ≈сли обратитьс€ к схематической моде≠ли полифункционального субстрата 156 (схема 2.73), то можно ожидать, что все три показанные функциональные группы будут способны восстанавли≠ватьс€ этим реагентом. ќднако известно, что эти группы достаточно заметно отличаютс€ по реакционной способности и могут быть расположены в сле≠дующий р€д по мере уменьшени€ электрофильности: —Ќќ > —ќќће > —Ќ2—1. Ёто про€вл€етс€, в частности, в том, что легко и быстро протекают реакции алюмогидрида лити€ с альдегидной и сложноэфирной функци€ми, но не с первичными хлоридами. ѕоэтому получение хлордиола 157 Ч триви≠альна€ задача.
¬ то же врем€ селективное восстановление альдегидной группы в 156 осу≠ществить с помощью того же реагента не удастс€, так как будет протекать также восстановление сложноэфирной группы, ибо скорости этих двух реак≠ций, хот€ и различимы, но вполне сопоставимы. ≈сли бы алюмогидрид был единственным доступным источником гидрид-иона, то эффективное общее решение этой задачи найти было бы трудно. ќднако существует широкий на≠бор гидридных восстановителей, аналогичных по типу алюмогидриду, но значительно отличающихс€ от него как по активности, так и по другим ха≠рактеристикам [23а]. ќдин из них Ч это боргидрид натри€. —в€зь ¬-Ќ в этом соединении несколько прочнее св€зи јl-Ќ, и этот реагент €вл€етс€ более слабым нуклеофилом по сравнению с алюмогидридом. Ѕлагодар€ этому раз≠личи€ в скорост€х реакций боргидрида с альдегидной и сложноэфирной группами €вл€ютс€ препаративно значимыми, и поэтому с помощью бор-гидрида восстановление альдегидной группы в полифункциональной систе≠ме типа 156 вполне может быть осуществлено хемоспецифично с исключи≠тельным образованием продукта 158.
 —хема 2.73
—хема 2.73
|
Ќаконец, если задача состоит в исчерпывающем восстановлении субстра-татипа 156, то можно воспользоватьс€ еще одним, на этот раз более мощным донором гидрид-иона. –ечь идет диборане ¬2Ќ6, о котором мы уже упомина≠ли в св€зи с гидроборированием. ƒиборан успешно атакует (в несколько бо≠лее жестких услови€х) даже относительно слабые электрофилы типа хлорме-тепьной группы в 156, и с его помощью этот субстрат может быть превращен в диол 159. ≈ще более эффективно подобное исчерпывающее восстановле≠ние- может быть проведено с помощью так называемого супергидрида LiEt3BH, €вл€ющегос€ одним из самых мощных нуклеофилов [191]. ѕара≠доксально, но и обратна€ задача селективности, а именно восстановление самой слабой по электрофильности хлорметильной группы в присутствии альдегидной или сложноэфирной, также может быть решена за счет выбора ѕодход€щего гидридного восстановител€, которым на этот раз оказалс€ цианоборгидрид натри€ NaBH3CN [23b].
|
|
|
ћетоды восстановлени€ комплексными гидридами с использованием самых разнообразных (но в принципе однотипных!) реагентов разработаны сейчас настолько хорошо, что задачу хемоселективного восстановлени€ жить одной из групп возможно решить при почти любой комбинации вос≠станавливаемых функций в субстрате. “ак, например, хорошо известна способность а,–-непредельных альдегидов и кетонов в реакци€х с восста≠новител€ми самых различных типов (например, боргидридом натри€) об≠разовывать смеси продуктов как 1,2-, так и 1,4-присоединени€. ќднако мо≠дификацией природы гидридного восстановител€ можно обеспечить лю≠бую хемоселективность при проведении этой реакции. “ак, чистое 1,2-вос-—“ановление может быть проведено с использованием системы NaBRj/CeCb [23c| или Zn(BH4)2 [23d] (схема 2.74). »нтересно, что с по≠мощью последнего реагента удаетс€ также провести селективное восста≠новление изолированной кетогруппы в присутствии сопр€женной еноновой системы [23е].
—равнительно недавно арсенал сингетиков обогатилс€ за счет по€влени€ нового семейства гидридных восстановителей, литийаминоборанов LiABH3 (ј Ч циклический амин) [23f]- Ёти реагенты оказались исключительно ак≠тивными и селективными в 1,2-восстановлении сопр€женных альдегидов и кетонов, а их дополнительным преимуществом, выгодно отличающим их от самого алюмогидрида лити€, €вл€етс€ мала€ чувствительность к влаге.
 —хема 2.74
—хема 2.74
|
≈сли же синтетическа€ задача требует, напротив, селективного 1,4-воста-новлени€ еноновой системы, то в качестве реагента дл€ проведени€ этого превращени€ может использоватьс€ алюмогидрид лити€ в присутствии ком≠плексов меди [23g] или такой кчассический восстановитель, как литий в жидком аммиаке [23h]. Ќаконец, исчерпывающее восстановление еноново-го фрагмента может быть осуществлено с помощью мощного гидридного восстановител€ ¬Ќ(втoр -¬u)3 или, в некоторых случа€х, в услови€х ионно≠го гидрировани€ [23j].
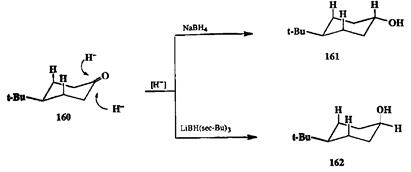
Ќа примере восстановлени€ комплексными гидридами удобно показать также возможности управлени€ и другим важнейшим параметром селективно≠сти, а именно стереоселекгивностью восстановлени€. ак мы уже упоминали в начале этой главы, восстановление 4- трет -бутилциклогексанона (160) мо≠жет приводить к образованию смеси двух изомерных спиртов, 161 и 162 (схе≠ма 2.69). ≈сли в качестве реагента дл€ восстановлени€ использовать NaBH4, то основным продуктом €вл€етс€ термодинамически более стабильный транс- изомер 161 (схема 2.75). ≈сли, однако, проводить восстановление с помощью гидридного реагента, содержащего объемистые группы, как, например, 1л¬Ќ(Ђ€ор-¬и)з, то селективность реакции мен€етс€ на обратную и основным продуктом оказываетс€ г<мс-изомер 162 [23i]. Ќаблюдаема€ зависимость, как полагают, обусловлена тем, что наличие в субстрате 160 двух аксиальных водородов при —-3 и —-5 эффективно блокирует подход объемистого реагента LiBH(втор -¬и)3к карбонильной группе Ђсверхуї от плоскости цикла, но в существенно меньшей степени преп€тствует такому направлению атаки дл€ реагента меньшего размера, каким €вл€етс€ NaBH4- ѕарадоксально, но уже упоминавшавшиес€ выше литийаминобораны ведут себ€ как стерически незатрудненные реагенты и независимо от размера аминной группы дают практически индивидуальный (99%) транс-изомер 161 [23f].
|
|
|
ѕодобный описанному принцип Ђдозированного воздействи€ї Ч применени€ однотипных, но различных по определенным характеристикам реа-гентов дл€ обеспечени€ требуемого характера селективности Ч широко ис≠пользуетс€ в современном органическом синтезе. Ѕолее того, можно утверж≠дать, что без наличи€ подобного многообрази€ синтетических инструментов, искусно настроенных на решение той или иной из конкретных задач селек≠тивности, вообще невозможно было бы осуществление очень многих из со≠временных полных синтезов.
Ќе менее важно наличие широкого набора реагентов дл€ тех или иных гетеролитических реакций образовани€ св€зи —-—. ¬ этой области, пожа≠луй, наибольшее разнообразие характерно дл€ нуклеофильных реагентов. “ак,известны дес€тки типов металлоорганических реагентов, которые со≠держат один и тот же органический остаток и различаютс€ лишь природой металла и св€занных с ним лигандов [4]. ѕодобные, в сущности очень сход≠ные реагенты, разработанные дл€ сочетани€ одного и того же нуклеофиль-»ого остатка с электрофильными реагентами, на самом деле могут значи≠тельно различатьс€ по своей нуклеофильности, основности, способности к комплексообразованию и т. д. Ѕлагодар€ этому можно решительным обра≠зом вли€ть на селективность реакций образовани€ св€зи —-— в применении к взаимодействию как с электрофильными субстратами разных типов, так и с полидентатными электрофилами. “ак, взаимодействие классических реа≠гентов √ринь€ра со сложными эфирами или хлорангидридами не может быть остановлено на стадии образовани€ кетона, и продуктами такой реак≠ции неизменно €вл€ютс€ третичные спирты. ¬ то же врем€ замена магниевых производных на производные кадми€ [24а] или марганца [24№] делает реакцию с хлорангидридами удобным методом синтеза несимметричных кстонов. Ќа основе последовательности реакций хлорангидридов, первона≠чально с марганецорганическими производными, а затем с магнийорганическими реагентами, удалось разработать простой способ получени€ несим≠метричных третичных спиртов по схеме сборки из трех предшественников в одном реакционном сосуде [24с].
 —хема 2.75
—хема 2.75
|

ќсобое место среди всего многообрази€ классов и типов металлооргани≠ческих реагентов принадлежит медьорганическим производным. ’от€ мы уже неоднократна упоминали об их использовании в роли синтетических эквивалентов карбанионов в таких реакци€х образовани€ св€зи —Ч—, как со≠четание по ¬юрцу или присоединение по ћихаэлю, здесь уместно несколь≠ко более подробно рассмотреть специфику использовани€ этих реагентов, но на этот раз с точки зрени€ селективности превращений, обеспечиваемых их участием в некоторых реакци€х образовани€ св€зи —-—.
ћедьорганические соединени€ типа RCu, как таковые, не очень часто примен€ютс€ в синтетически значимых превращени€х за исключением не≠которых специфических случаев синтеза ацетиленовых соединений. Ќапро≠тив, смешанные купраты переменного состава от R2CuLi до R3Cu2Li [24d|, a также их всевозможные комплексы с лигандами, такими, как Me:S, Ph3P, RS~, CN~ и т. д. [15с,d], наход€т самое разнообразное применение. этому же типу реагентов относ€тс€ и магаийорганические реагенты, модифициро≠ванные добавками солей меди и лигандов, например, RMgBr/CuBr/Me2S.
»нтересно хот€ бы гасратце познакомитьс€ с историей разработки куп-ратных реагентов. ѕервое медьорганическое производное, димегилмедь (ће2—и), было получено √илманом в 1936 г. [24е]. ÷елью этой работы €вл€≠лось просто расширение круга известных в то врем€ металлоорганических соединений и изучение их свойств вне какой-либо св€зи с общими пробле≠мами органического синтеза. Ќесколько позднее (в 1941 г.) довольно случай≠но было обнаружено, что реакционна€ способность классических реагентов √ринь€ра может измен€тьс€ в присутствии неорганических солей, в частно≠сти солей меди. ƒолгое врем€ эти результаты казались частност€ми и поэто≠му не привлекали особого внимани€. ќднако ситуаци€ изменилась реши≠тельным образом в 1960-х годах, когда в св€зи с задачей синтеза феромонов и простаноидов возникла остра€ необходимость в разработке общих и препа≠ративно приемлемых методов управлени€ селективностью реакций карбани-онных нуклеофилов с полифункциональными электрофилами. ¬ св€зи с эгим вновь возник интерес к медьорганическим соединени€м, и вскоре бы≠ла показана перспективность использовани€ реагентов на их основе дл€ ре≠шение многих проблем селективности.
|
|
|
“ак, в работах ’ауса было найдено, что диметиллитийкупрат (Me2LiCu), реагент, полученный впервые еще в 1952 г. в лаборатории √илмана, про€вл€≠ет уникальную способность реагировать с а,р-непредельными альдегидами и кетонами с исключительным образованием продуктов сопр€женного 1,4-присоединени€ [24f,g]. јналогичным образом реагировали и другие алхил-литийкупраты [24h]. Ёти результаты послужили мощным стимулом дл€ по≠следующих интенсивных исследований р€да групп, результатом которых €вилась создание обширного нового класса нуклеофилов Ч купратных реа≠гентов, эквивалентов карбанионов различной структуры. ”местно отметить, что все эти реагенты легко могут быть получены из обычных литий- или магнийорганических соединений путем добавлени€ требуемого количества со≠лей меди и модифицирующих добавок.
Ѕлагодар€ этим разработкам впервые удалось создать надежный и общий метод 1,4-присоединени€ —-нуклеофилов по двойной св€зи о,(3-непредель-ных карбонильных соединений (о стратегической важности этого метода, см. выше разд. 2.2.3.3, а также разд. 3.2.7) [15с]. эгому следует добавить, что если модифицировать те же самые реагенты √ринь€ра, но не сол€ми меди, а сол€ми цери€ (церийорганические реагенты), то с этими же субстратами можно столь же чисто провести исключительное 1,2-присоединение. “аким образом, за счет модификации исходного металлоорганичееского реагента тем или иным из упом€нутых способов удаетс€ эффективно управл€ть селек≠тивностью присоединени€ —-нуклеофилов по одному из двух электрофиль-ных центров, имеющихс€ в молекуле исходного сопр€женного карбониль≠ного соединени€ [4, 15b-d] (схема 2.76).
 —хема 2.76
—хема 2.76
|
ќ том, как с помощью все тех же купратных реагентов стало возможным проводить селективное сочетание по схеме реакции ¬юрца, мы уже говори≠ли выше (см. разд. 2.2.3.1). ¬ этой св€зи необходимо также сказать еще о таких синтетически значимых особенност€х свойств купратов, как их сравни≠тельна€ инертность по отношению к карбонильным электрофилам и, напро≠тив, необычно высока€ активность по отношению к таким слабым электро≠филам, как винил- или арилгалогениды [24ij]. Ёти особенности купратов позвол€ют использовать эти нуклеофилы как реагенты дл€ селективного ал-килировани€ полифункциональных электрофилов различных типов, как это показано на модельных примерах на схеме 2.77.
 —хема 2.77
—хема 2.77
|
“ема вариабильности свойств нуклеофилов карбанионного типа в зави≠симости от природы металла получила интересное развитие в работах нохел€ [24k]. ’орошо известно, что цинкорганические реагенты относ€тс€ к ка≠тегории довольно малоактивных нуклеофилов. »сследовани€ми группы нохел€ было показано, что активность этих реагентов существенно возра≠стает, если их модифицировать добавлением цианида меди. јвторы разрабо≠тали метод, позвол€ющий превращать дииодалканы типа 163 (схема 2.78) в гетеробиметаллические производные типа 163а. Ќаличие в последних двух достаточно различных по активности нуклеофильных центров позволило провести с высокой селективностью два последовательных сочетани€ с раз≠личными электрофильными реагентами, как это представлено на схеме 2.78. —интетический интерес подобного тандема реакций представл€етс€ несом≠ненным.
|
|
|
 —хема 2.78
—хема 2.78
|
ƒо сих пор мы рассматривали способы регулировани€ селективности об≠разовани€ св€зей —Ч—, основанные на изменени€х в свойствах нуклеофиль-ной компоненты. ќчевидно, что не менее эффективными средствами такого контрол€ могут быть вариации в природе реагентов, эквивалентных одному и тому же электрофилу. Ќапример, столь различные по свойствам соедине≠ни€, как RCO+BF4-, RCOCl, (RCO)2O, RCOOR1, в реакци€х с нуклеофила-ми выступают в роли переносчиков одного и того же ацил-катиона. “очно также такие непохожие соединени€, как соли триалкилоксони€ R3O+BF4-, алкилтозилаты, алкилгалогениды или алкилацетаты, могут использоватьс€ в качестве эквивалентов одного и того же алкил-катиона. ѕон€тно, что при наличии столь богатого арсенала электрофильных реагентов, различающих≠с€ по своей активности, стабильности, чувствительности к стерическим пре-п€тстви€м, эффектам растворител€ и т. п., почти всегда можно выбрать такой реагент, который обеспечит нужную хемо- или региоселективность реакции с субстратом, имеющим несколько нуклеофильных центров.
¬есьма поучительным примером того, насколько может быть эффектив≠ной регулировка селективности за счет казалось бы не очень значительных вариаций в природе электрофильной и/или нуклеофильной компонент, мо-жет служить результаты исследований группы отсуки [241], направленных наразработку общей методологии синтеза р€да структурно различных энан-тиомерно чистых феромонов (см. схему 2.79).
 —хема 2.79
—хема 2.79
|
«амысел работы заключалс€ в разработке способа использовани€ легко доступного в энантиомерно чистом виде 2,3-ќ-изопропилиден-D-трент 164 в качестве предшественника бифункционального электрофила, который да-леепредполагалось вводить в реакции последовательного контролируемого сочетани€ с набором нуклеофилов. ƒл€ достижени€ этой цели 164 был пре≠вращен в смешанный тозилат-трифлат 165, что обеспечило создание двух электрофильных центров, различающихс€ по своей активности. ƒалее требовалось найти нуклеофилъные реагенты, способные селективно реагиро≠вать по одному из этих элсктрофильных центров. ќказалось, что стандарт≠ные диалкиллитийкупратные реагенты Ђне чувствуютї различи€ в активно≠сти тозилатной и трифлатной групп, и оба этих центра подвергались нуклео-фильнай атаке примерно с одинаковой легкостью. Ќапротив, менее актив≠ные реагенты алкилмагнийкупратного типа не реагировали с тозилатной функцией и замещению подвергалась исключительно трифлатна€ группа. ѕолучающийс€ на первой стадии продукты далее повторно алкилировали, на этот раз по оставшейс€ тозилатной группе, с помощью диалкиллитий-купратных реагентов. ѕримечательно, что обе стадии алкилировани€ про≠вод€тс€ как последовательность реакций в одном реакционном сосуде и привод€т к образованию целевых продуктов с высоким выходом. –азрабо≠танна€ схема несимметричного бис-алкилировани€ допускает возможность независимого варьировани€ строени€ нуклеофильного остатка в реагентах, используемых на первой и второй стади€х последовательности (см., напри≠мер, получение аддуктов 166 и 167, схема 2.79). “аким образом, на основе общего хирального бифункционального субстрата 165 можно получать ши≠рокий набор разнообразных продуктов, которые далее могут использовать≠с€ как субстраты в синтезе энантимерно чистых целевых соединений, в том числе и р€да феромонов. ќтметим еще раз, что успешное решение подобно≠го рода непростых задач стало достижимым только благодар€ наличию ши≠роких возможностей тонко дозируемых изменений свойств примен€емых реагентов.
—елективна€ активаци€

