




САНКТ-ПЕТЕРБУРГСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
Направление подготовки: Химия
Образовательная программа: Химия
КУРСОВАЯ РАБОТА
Реакция Вильгеродта-Киндлера
Студент 3курса 1 группы
А. Д. Герасимова
Уровень/ступень образования:
Бакалавриат
Научный руководитель:
к.х.н., Е. Е. Галенко
Санкт-Петербург
2018 год
Содержание
1. Введение………………………………………………………………………….……3
2. Литературный обзор…………………………………………………………………..4
2.1. Реакция Вильгеродта – Киндлера………………………………………………….4
2.2. Ацилирование по Фриделю-Крафтсу……………………………………………...7
3. Экспериментальная часть…………………………………………………………….9
Заключение……………………………………………………………………………...11
Список литературы……………………………………………………………………..12
Введение
Для получения арилалифатических карбоновых кислот из доступных алкиларилкетонов применяют реакцию, открытую в 1887 году Конрадом Вильгеродтом. Вильгеродт открыл реакцию образования фенилацетамида при нагревании ацетофенона с водным раствором полисульфида аммония. Позже эта реакция была проведена с различными арилкетонами. Во всех случаях карбонильная группа восстанавливается до метиленовой, а на конце цепи образуется амидная группа, после чего гидролизом может быть получена карбоновая кислота. Все эти реакции получили название реакции Вильгеродта [1]. Киндлер установил, что вместо водного раствора полисульфида аммония можно использовать серу и сухие амиды. При этом в качестве главного продукта реакции образуются тиоамиды [2].
Литературный обзор

Реакция Вильгеродта-Киндлера
Реакция Вильгеродта-Киндлера известна с конца 19 века. Механизм реакции изучался многими авторами и был предметом длительной дискуссии. Опытами, проведенными с кетонами, молекулы которых содержат меченный углерод, установлено, что реакция не сопровождается перегруппировкой углеродного скелета. Доказано, что при превращении продукта реакции в бензиламин радиоактивность сохраняется:
[3].
Атакующей частицей в данной реакции является продукт взаимодействия серы с жидким аммиаком, первичным или вторичным амином:
R 2 NH + S 8 → R 2 N +∙ S -∙ 8 [1].

Далее встает вопрос о том, какое место в кетоне подвергается атакой радикалами. Если предположить, что атака идет по углероду карбонильной группы, то реакция должна протекать с образованием амина или сульфида. Действительно, некоторые опыты подтверждают эту теорию, тогда механизм реакции выглядит следующим образом:

Однако при введении в реакцию пространственно замещенных кетонов карбонильная группа не затрагивается, и образуется тиоморфолид:
Эта реакция идет не по карбонильной группе, а начинается с окисления стоящей в α-положении метиленовой группы.
Для объяснения процесса окисления метиленовой группы было предложено три различных механизма:
Аминный механизм

Механизм предложен Кармаком, и считается маловероятным. Он включает в себя стадии «присоединение амина – отщепление воды». В основе его лежит присоединение на промежуточной стадии амина и последующее образование тройной связи:
Этот механизм не может объяснить протекание реакции даже для таких соединений, как изобутилфенилкетон, для которых образование тройной связи в ходе реакции невозможно:

Существует еще несколько опровержений представленного механизма, однако нельзя полностью исключать его из рассмотрения. Первая часть механизма, образование енамина, может иметь место, дальнейшее же течение реакции может включать стадии присоединения полисульфидного ион-радикала к полученному енамину, отщепления амина и изомеризации:

Тиольный механизм

Данный механизм был предложен Кингом и Мак Милланом. Согласно ему, в начале происходит восстановление карбонильной группы до тиольной, далее эта группа отщепляется, что приводит к образованию алкена, к которому присоединяются серный и аминный радикалы:
Тиоэпоксидный механизм
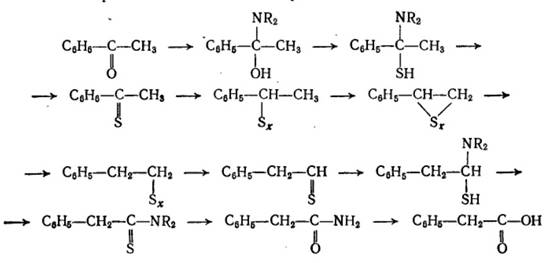
Этот механизм был предложен Тогава. В роли окислителя кетона здесь выступает сера:
Однако, представленные механизмы не способны объяснить все известные случаи протекания реакции. Так, они не подходят для реакций соединений, не имеющих концевой метильной группы.
Примером такой реакции может быть равновесие, наблюдаемое в условиях реакции Вильгеродта между 1,3-дифенилпропаноном-2 и 1,3-дифенилпропаноном-1, в то время как соединения, которые могли бы служить промежуточными в этом превращении: тиол, алкин или алкен, в данных условиях не переходят в карбонильные соединения.

Следовательно, изомеризация карбонильных соединений не обязательно осуществляется через образование алкенов, тиолов или алеинов в качестве промежуточных соединений, а предложенные механизмы не дают исчерпывающего описания протекания всех известных случаев реакции [1].
В литературе также описывается механизм реакции Вильгеродта, несколько отличный от данных выше, представляющий собой ряд последовательных отщеплений и присоединений элементов сероводорода, воды или амина, с образованием промежуточных ненасыщенных соединений:
RCOCH2CH3→RCSCH2CH3→R-CH(SH)-CH2CH3→R-CH=CH-CH3→RCH2-CH(SH)-CH3→RCH2CH=CH2→RCH2CH2CH2SH→RCH2CH2CH=S→RCH2CH2CONH2
Ацилирование по Фриделю-Крафтсу
Арилалифатические кислоты по реакции Вильгеродта получаются из доступных алкиларилкетонов, получаемых по реакции Фриделя-Крафтса. Эта реакция является одним из самых распространенных методов синтеза арилкетонов. В качестве ацилирующих агентов используются хлорангидриды и ангидриды кислот, что позволяет ацилировать даже слабо дезактивированные арены. В качестве катализатора добавляют стехеометрическое количество галогенида металла – обычно безводного хлористого алюминия [4]. При протекании реакции хлорид алюминия координируется по атому кислорода ацильной группы, образуя ацилирующий агент – комплекс, которые далее вступает в реакцию 
электрофильного замещения с ареном, тяготея к пара-положению [5].
Наилучшие выходы синтеза карбонильных кислот из арилкетонов наблюдаются для арилуксусных кислот, в сравнении с гомологами. Чисто алифатические кетоны также могут вступать в реакцию Вильгеродта, но они дают более низкие выходы, причем выход тем ниже, чем дальше отстоит карбонильная группа от конца цепи[3].
Экспериментальная часть
Синтез 4 ’ -гексилоксиацетофенона

В колбу, снабженную магнитной мешалкой, обратным холодильником и капельной воронкой, было помещено 48 г (0,36 моль) безводного хлористого алюминия и 200 мл безводного четыреххлористого углерода. При перемешивании добавлено по каплям 26 г (0,33 моль) хлорангидрида уксусной кислоты, затем 53,4 г (0,3 моль) гексилоксибензола. Смесь перемешивалась при комнатной температуре в течение 2 часов, до прекращения выделения хлороводорода. Содержимое колбы было перелито в стакан, содержащий смесь льда и 450 мл 2 N раствора соляной кислоты. Органический слой был отделен и промыт водой. Растворитель отогнали, остаток перегнали в вакууме. Выход кетона составил 32, 5 г - 52 %.
Синтез 4 ’ -гексилоксифенилуксусной кислоты

В колбу, снабженную магнитной мешалкой и обратным холодильником, поместили 32,5 г (0,15 моль) 4’гексилоксиацетофенона, 26 мл морфолина, 10 г тонкорастертой серы и 50 мл ДМФА. Реакционную смесь перемешивали при нагревании на кипящей водяной бане в течение 4 часов. Затем вылили в стакан с холодной водой. Выпавший осадок тиоморфолида отфильтровали на воронке Бюхнера, промыли 60 мл воды и просушили на листе фильтровальной бумаги.
Неочищенный тиоморфолид и 25 г 50% раствора КOH в 50 мл этилового спирта кипятили в течение 6 часов с обратным холодильником. Большую часть спирта отогнали, остаток разбавили 15 мл воды, отфильтровали и подкислили соляной кислотой до рН 1-2. Выпавшую при охлаждении кислоту отфильтровали на воронце Бюхнера и промыли водой. Выход кислоты составил 8,9 г - 25%.
Заключение
Были рассмотрены теоретические аспекты реакций Вильгеродта – Киндлера и ацилирования по Фриделю – Крафтсу, и проведён контрольный синтез с участием гексилоксибензола. Выход 4’-гексилоксифенилуксусной кислоты составил 13%.
Литература
[1]. Сигэру Оаэ, «Химия органических соединений серы», пер. с яп. к. х. н. Ян Юн Бина и к. х. н. Б. К. Нефедова, Москва, изд. «Химия», 1975
[2]. А. Серрей, «Справочник по органическим реакциям. Именные реакции в органической химии», пер. с англ. д. х. н. Н. С. Вульфсона, Государственное научно-техническое издательство химической литературы, Москва, 1962
[3]. Краткая химическая энциклопедия под ред. И. Л. Кнунянц, т. 1, «советская энциклопедия», Москва, 1961
[4]. Костиков Р. Р., Кузнецов М. А., Новиков М. С., Соколов В. В., Хлебников А. Ф., Практикум по органическому синтезу, Санкт-Петербург, 2009
[5]. Агрономов А. Е., «Избранные главы органической химии», изд. «Химия», Москва, 1990

