




Рис.1.1

Рис.1.2

Рис.1.3

РАЗДЕЛ II
АНЕМИИ
Анемия – это синдром, либо самостоятельное заболевание, характеризующееся понижением содержания в крови гемоглобина, которое как правило сопровождается понижением содержания эритроцитов. Клиническая симптоматика характеризуется общими признаками для всех видов анемий: головокружение, шум в ушах, слабость, обмороки. Анемиями страдает примерно от 10 до 20% всего населения земли, из них 30% женщины и 50% дети.
Классификация анемий:
1. По типу эритропоэза выделяет нормобластические и мегалобластические анемии.
2. По диаметру эритроцитов выделяют нормоцитарные анемии (7,5-8,5 мкм); макроцитарные анемии (более 8,5 мкм); микроцитарные анемии (менее 7,5 мкм).
3. По цветовому показателю выделяют нормохромные анемии (0,85-1,05), гипохромные (менее 0,85) и гиперхромные (более 1,05).
Цветовой показатель рассчитывается по формуле: 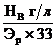 .
.
4. По регенераторной способности красного костного мозга выделяют: регенераторные анемии – 0,8-10% ретикулоцитов, гиперрегенаторные – более 10% ретикулоцитов и гипорегенераторные – менее 10% ретикулоцитов. В норме содержание ретикулоцитов составляет от 0,2 до 1%.
5. По этиопатогенезу выделяют анемии:
а)вследствии кровопотери. К ним относятся острая постгеморрагическая и хроническая постгеморрагическая анемии.
б)вследствии нарушенного кровообразования: железодефицитная, В12 и фолиеводефицитная, гипо- и апластическая анемии.
в)вследствии избыточного кроворазрушения: наследственные и приобретенные гемолитические анемии.
Практически при всех видах анемий имеет место качественное изменение со стороны эритроцитов. К ним относятся: анизоцитоз, пойкилоцитоз, анизохромия, наличие патологических включений в эритроцитах (тельца Жолли и кольца Кебота), базофильная пунктация, в следствии коагуляции белков цитоплазмы, сидероциты и сидеробласты.
Характеристика отдельных видов анемий.
Острая постгеморрагическая анемия развивается при одномоментной кровопотери более 10% объема циркулирующей крови. Этиология: травмы, внематочная беременность с разрывом трубы, гемофилия. В развитии ОПГА выделяют три фазы:
1-я фаза – кратковременный спазм, обусловлен выбросом катехоламинов.
2-я фаза – гидремической компенсации. Характеризуется увеличением ОЦК в результате задержки жидкости в кровеносном русле. Это происходит в результате активации ренин – ангиотензиновой системы. Схема которой следующая: ишемия юкстагломерулярного аппарата почек ® выброс ренина ®--> активация ангиотензиногена и превращение его в ангиотензин-1, превращение ангиотензина I в ангиотензин-2 под действием АПФ ® усиление синтеза альдостерона ® задержка натрия® усиление выброса антидиуретического гормона ® задержка воды ® увеличение ОЦК.
3-я фаза – костномозговой компенсации, характеризуется усилением выработки эритропоэтинов в почках и активацией костномозгового кроветворения. Эта фаза формируется примерно через неделю после кровопотери. Полное восстановление картины крови происходит спустя 4-6 недель.
Гематологическая характеристика картины крови при ОПГА: анемия нормобластическая, нормоцитарная, нормо- или легкая гипохромная, регенераторная, с умеренным анизо и пойкилоцитозом.
Железодефицитная анемия. Характеризуется истощением депо железа в организме. В организме взрослого человека содержится от 4 до 5 грамм железа. При этом 70% приходится на долю гемоглобина, 25% на долю ферритина и гемосидерина, 4% на долю миоглобина и 1% на долю цитохромов.
Этиология:
1. Недостаточное поступление железа с пищей. В норме с пищей поступает 15-20 мг. железа, но всасывается из них только 1-2 мг. Полностью усваивается железо содержащееся в мясе, так как оно двухвалентное. Железо содержащееся во фруктах может усваиваться только с аскорбиновой кислотой, так как оно трехвалентное.
2. Недостаточное всасывание железа из кишечника.
3. Дефицит трансферрина (наследственный или приобретенный).
4. Избыточное потребление железа плодом при беременности.
5. Хронические кровопотери.
6. Избыточное поглощение железа гельминтами.
Более подробно причины дефицита железа будут рассмотрены ниже.
Ежедневно для синтеза гемоглобина требуется примерно 25 мг железа. 1 мг поступает с пищей и 24 мг поступает из депо. Развитию гематологической картины железодефицитной анемии предшествует железодефицитное состояние, которое характеризуется снижением содержания сывороточного железа. Основным клиническим проявлением железодефицитного состояния является пикохлоротика, которая проявляется извращением вкуса и обоняния.
Клиника железодефицитной анемии. Для железодефицитной анемии характерны следующие клинические синдромы: гемический, трофических нарушений, иммунодефицитное состояние.
Гематологическая характеристика: нормобластическая, микроцитарная, гипохромная, гипорегенаторная анемия, сопровождающаяся лейкопенией, пониженным содержанием в крови сывороточного железа (норма 9-31 мкм/л) и увеличением общей железосвязывающей способности крови за счет увеличенного содержания свободного трансферрина (норма 45-75 мкм/л).
Разновидностью железодефицитной анемии является сидероахристическая анемия, при которой нарушается включение железа в молекулу гема. Эта анемия характеризуется повышенным содержанием сывороточного железа и снижением общей железосвязывающей способности крови.
Картина крови и костного мозга
В мазке крови обнаруживают гипохромию и микроцитоз, а также пойкилоциты (обычно в виде сигары) и иногда – мишеневидные эритроциты (см. 2.1). Выраженность изменений в мазке крови, а также степень снижения среднего эритроцитарного объема и среднего содержания гемоглобина в эритроците соответствует тяжести анемии. Число тромбоцитов обычно повышено, особенно при кровопотере.
Костный мозг характеризуется нормальной клеточностью, в ряде случаев наблюдается гиперплазия эритроидного ростка. Для предшественников эритроцитов характерны неровные контуры и вакуолизация цитоплазмы. Окраска по Перльсу выявляет полное отсутствие запасов железа, гранулы гемосидерина в эритрокариоцитах отсутствуют.
Причины дефицита железа
Остановимся более подробно на причинах дефицита железа. Примерно две трети железа в организме входит в состав гемоглобина: каждый литр крови содержит приблизительно 500 мг железа. Второй по величине резерв – до 2 г- это запасы железа в макрофагах в виде гемосидерина (определяется при световой микроскопии) и ферритина (определяется только при электронной микроскопии). Состояние, когда запасы железа отсутствуют, уровни сывороточного железа и ферритина снижены, общая железосвязывающая способность сыворотки повышена, но анемии нет и эритроцитарные индексы в норме, называется латентным дефицитом железа. Остальное железо организма находится в составе миоглобина и различных ферментов.
Потери железа по сравнению с его запасами в организме у взрослого человека в норме незначительны. Следовательно, невелики и потребности в нем: они составляют примерно 1 мг в сутки у мужчин и женщин в постменопаузе и несколько больше – 1,5-3,9 мг в сутки – у женщин детородного возраста. Потребности в железе повышены у детей (вследствие активного роста и увеличения объема циркулирующих эритроцитов) и у беременных (из-за потребления железа плодом).
Дефицит железа, как правило, обусловлен хронической кровопотерей. Самой частой ее причиной во многих странах служат анкилостомидозы (см. 2.2), при этом объем кровопотери зависит от численности паразитов. Распространенные причины хронической кровопотери у женщин – обильные продолжительные менструации и частые беременности без приема препаратов железа. У женщин в постменопаузе и мужчин дефицит железа обычно обусловлен повторяющимися желудочно-кишечными кровотечениями, основными причинами которых в западных странах служат грыжа пищеводного отверстия диафрагмы, язвенная болезнь, длительный прием аспирина, рак ободочной и слепой кишки, ангиодисплазия, дивертикулез толстой кишки, геморрой. Редкие причины дефицита железа – идиопатический гемосидероз легких, хронический внутрисосудистый гемолиз (например, при пароксизмальной ночной гемоглобинурии) и самостоятельные кровопускания.
Суточный рацион жителей западных стран обычно содержит 10-15 мг железа, из которых всасывается 5-10%. Всасывание железа усиливается при его дефиците, но тормозится такими компонентами пищи, как фитаты и фосфаты. Низкое поступление железа с пищей может стать единственной причиной дефицита железа, если оно сохраняется на протяжении многих лет. Однако гораздо чаще недостаточное потребление железа лишь уменьшает его запасы в организме, создавая тем самым условия для возникновения дефицита железа и анемии под действием других факторов (менструальных кровопотерь, беременности, ускоренного роста у детей).
Нарушение всасывания само по себе – тоже редкая причина дефицита железа. При атрофическом гастрите и целиакии железо теряется за счет усиленного распада клеток и выведения трансферрина, и эти потери по значимости сопоставимы с теми, которые вызваны нарушением его всасывания. После резекции желудка дефицит железа развивается вследствие двух основных причин- кровопотери и нарушения всасывания; причем страдает главным образом всасывание органических соединений железа, а не неорганических.
Лечение дефицита железа преследует две цели. Первая из них – установить причину дефицита и по возможности ее устранить. Вторая – назначить препараты железа в дозе, необходимой для излечения анемии и восстановления запасов железа в тканях. Лучше всего использовать препараты для приема внутрь, однако изредка требуется парентеральное введение препаратов железа, например одномоментное введение комплекса декстран – железо в восполняющей дефицит дозе. При правильном лечении уровень гемоглобина возрастает на 2 г% каждые 3 нед.
Эритропоэтические порфирии
Для эритропоэтической уропорфирии и эритропоэтической протопорфирии – наследственных нарушений синтеза порфиринов с повышенной чувствительностью к солнечному свету – характерно поражение системы кроветворения.
Эритропоэтическая уропорфирия
Эритропоэтическая уропрфирия (болезнь Гюнтера, или врожденная эритропоэтическая порфирия) наследуется аутосомно-рецессивно и характеризуется избыточной продукцией уропорфириногена I, из которого образуется уропорфирин I и копропорфирин I. В основе нарушений лежит недостаточность уропорифириноген-III-синтазы – фермента, участвующего в синтезе гема. В плазме и эритроцитах в избытке накапливаются уропорфирин I, копропорфирин I и проторфирин.
У больного эритропоэтической уропорфирией наблюдаются везикулы и пузыри на открытых участках кожи, вскрывающиеся с образованием язв, гипертрихоз и гемолитическая анемия со спленомегалией. Моча красного цвета и светится под лампой Вуда. Кроме того, отмечается потемнение зубов и костей; для них также характерно свечение под лампой Вуда. Еще один признак заболевания – свечение эритробластов и нормобластов в ультрафиолетовом свете. При этом заболевании на коже под действием солнечных лучей тоже образуются пузыри. Недостаточность уропорфириногендекарбоксилазы, лежащая в его основе, бывает наследственной и приобретенной (например, в результате перегрузки железом, гиперэстрогении или злоупотребления алкоголем). Перегрузка железом может быть как следствием, так и причиной заболевания.
Эритропоэтическая протопорфирия
Эритропоэтическая протопорфирия наследуется аутосомно-доминантно. В основе заболевания лежит дефект феррохелатазы – фермента, катализирующего заключительную стадию синтеза гема. Образующийся в избытке протопорфирин накапливается в эритроцитах, печени и других тканях. Характерный признак заболевания – свечение эритрокариоцитов в ультрафиолетовом свете.
При эритропоэтической протопорфирии тоже повышена чувствительность к солнечному свету, наблюдаются зуд, отечность и покраснение кожи. Однако моча и зубы не изменяют окраску и не дают свечения. Не бывает и гемолитической анемии. Причиной гибели больного может быть печеночная недостаточность, развившаяся вследствие холестаза, гепатита и цирроза печени.
Перегрузка железом.
Гемохроматоз
Гемохроматозом называют наследственное нарушение обмена железа, характеризующееся усиленным всасыванием железа в кишечнике и накоплением его избытка в тканях. На сегодняшний день установлена генетическая природа большинства случаев гемохроматоза.
Клинические проявления гемохроматоза в значительной степени сходны с симптомами трансфузионного гемосидероза. Они включают гиперпигментацию кожи и эндокринные нарушения: сахарный диабет; дисфункцию половых желез, гипофиза, щитовидной и паращитовидных желез. Непременно происходит отложение железа в гепатоцитах, вследствие чего могут развиться фиброз, цирроз печени и печеночно-клеточный рак. Возможна кардиомиопатия с аритмией, перикардитом и сердечной недостаточностью. Асимметричный артрит, обычно дистальных межфаланговых суставов, с отложением кристаллов пирофосфата кальция характерен только для гемохроматоза и не встречается при трансфузионном гемосидерозе.
Известно, что ответственный за развитие гемохроматоза ген расположен на коротком плече 6-й хромосомы вблизи локуса HLA-A и тесно связан с локусом HLA-A3, в меньшей степени – с HLA-B7. Не так давно он был идентифицирован. Это ген HFE (известный также как HLA-H), кодирующий одноименный белок. Ген HFE, во многом сходный с генами HLA класса I, располагается на расстоянии 3 млн нуклеотидов от локуса HLA-A в направлении теломеры (Feder et al. Nat Genet, 13:399-408,1996). В нем содержится семь экзонов. По данным большинства исследований,83-100% больных гемохроматозом гомозиготны по миссенс-мутации, состоящей в замене гуанозина на аденозин в кодоне 845 и, соответственно, замене цистеина на тирозин в белке HFE в положении 282. Вторая из выявленных мутаций, заключающаяся в замене цитозина на гуанозин в экзоне 2, ведет к замене гистидина на аспарагиновую кислоту в положении 63. Незначительная доля больных оказалась смешанными гетерозиготами. Предполагаемая роль белка во всасывании железа продемонстрирована на рис.2.3. Есть сведения, что мутация гена HFE заставляет энтероциты двенадцатиперстной кишки функционировать, как при дефиците железа, усиленно синтезируя мембранный белок DMT1 (Divalent Metal Ion Transporter) – переносчик двухвалентных катионов. Это резко активизирует перенос железа из просвета кишки через щеточную каемку в энтероцит (см. 2.4). Ведение больных включает кровопускания для уменьшения перегрузки железом и симптоматическое лечение дисфункции пораженных органов.
Другие заболевания
Поздняя кожная порфирия сопровождается накоплением железа в печени. При редком аутосомно-рецессивном заболевании атрансферринемии наблюдается микроцитарная гипохромная анемия с избыточным отложением железа в клетках ретикулоэндотелиальной системы. Семейная ацерулоплазминемия, также наследуемая аутосомно-рецессивно, характеризуется дегенерацией сетчатки, поражением базальных ядер и накоплением железа в печени, поджелудочной железе, головном мозге и других органах. Уровень сывороточного железа низкий, общее содержание железа в организме в норме, уровень ферритина повышен. При атаксии Фридрейха дегенерации подвергаются спиноцеребеллярные пути. Заболевание проявляется абазией, атаксией и гипертрофической кардиомиопатией. Причиной служит мутация гена, кодирующего митохондриальный белок фратаксин. В сердце обнаруживают отложения железа.
Рис.2.1

Рис.2.2

Рис.2.3

Рис.2.4

Мегалобластные анемии
Мегалобластные анемии составляют группу анемий, для которых характерны макроцитоз и мегалобластный эритропоэз. Причины мегалобластных анемий представлены в табл.2.5. Как правило, причиной заболевания служит дефицит витамина В12 или фолиевой кислоты. В большинстве случаев дефектный этап в синтезе ДНК известен. Исключение составляют миелоидные лейкозы, миелодиспластические синдромы и некоторые другие заболевания, при которых мегалобластные изменения не поддаются лечению витамином В12 и фолиевой кислотой.
Роли витамина В12 и фолиевой кислоты в синтезе ДНК показаны на рис.2.6. При дефиците фолиевой кислоты тормозится образование тимидинмонофосфата (лимитирующая реакция синтеза пиримидиновых оснований), поскольку для него необходим полиглутамат 5,10- метилентетрагидрофолиевой кислоты, служащий коферментом. Коферментные формы фолиевой кислоты требуются также на двух этапах синтеза пуриновых оснований, но у человека эти реакции не относятся к лимитирующим синтез ДНК. Витамин В12 не участвует непосредственно в синтезе ДНК. Он необходим для превращения 5- метилтетрагидрофолиевой кислоты, поступающей в клетку из крови, в другие коферментные формы фолиевой кислоты (включая полиглутаматы). Этот процесс сопряжен с метилированием гомоцистеина до метионина под действием метионинсинтазы: 5-метилтетрагидрофолиевая кислота отдает метильную группу гомоцистеину и превращается в тетрагидрофолиевую кислоту, из которой путем присоединения остатков глутаминовой кислоты образуются полиглутаматы. Сама 5-метилтетрагидрофолиевая кислота не служит субстратом реакции образования полиглутамата. Роль фолиевой кислоты в обмене гомоцистеина показана на рис.2.7.
Содержащийся в пище витамин В12 под действием протеаз высвобождается из комплекса с белком и связывается с так называемым R-белком (см. 2.8). От него витамин В12 освобождается под действием ферментов поджелудочной железы, после чего соединяется с внутренним фактором Касла - белком, секретируемым обкладочными клетками желудка. В желчи витамин В12 также связан с внутренним фактором Касла. В комплексе с ним витамин В12 достигает повздошной кишки и связывается со специфическими рецепторами. Внутренний фактор Касла разрушается, а витамин В12 в комплексе с другим белком – транскобаламином II – поступает в кровь воротной вены. В крови витамин В12 в основном связан с транскобаламином I – гликопротеидом, источником которого служат гранулоциты, моноциты и их предшественники. Однако быстрое поступление витамина В12 в ткани обеспечивает транскобаламин II.
Фолаты пищи в эпителии тонкой кишки расщепляются до моноглутаматов, полностью восстанавливаются и метилируются, после чего всасываются в виде 5-метилтетрагидрофолиевой кислоты (см. 2.6).
Патогенез мегалобластных анемий.
Витамин В12 и фолиевая кислота являются донорами и акцепторами метильных групп и атомов водорода для ДНК и РНК. В результате при данных анемиях нарушается редупликация ДНК и синтез на ней РНК, как следствие угнетается митотический цикл всех быстро делящихся клеток, в том числе и клеток крови. Неврологический синдром обусловлен формированием фуникулярного миелоза, так как при недостатке витамина В12 нарушается превращение метилмалоновой кислоты в янтарную, как следствие – снижается синтез жирных кислот и нарушается миелинизация нервных волокон.
Клиническая картина
Мегалобластная анемия обычно развивается незаметно и прогрессирует настолько медленно, что организм вполне успевает приспособиться к возникшим изменениям. Следовательно, больной может не предъявлять никаких жалоб вплоть до развития тяжелой анемии, и заболевание нередко обнаруживают случайно, при анализе крови по другому поводу. Для больных с мегалобластными анемиями характерен лимонно-желтый оттенок кожи – результат сочетания желтухи и анемии. Желтуха обусловлена избыточным образованием непрямого билирубина из-за массовой гибели эритрокариоцитов в костном мозге (неэффективного эритропоэза) и внесосудистого гемоглобинолиза (идущего в клетках ретикулоэндотелиальной системы). Кроме того, вследствие усиленного разрушения эритроцитов значительно повышена активность ЛДГ в сыворотке; радиоактивное железо быстро выводится, лишь ничтожная его часть расходуется для эритропоэза.
В тяжелых случаях появляются признаки внутрисосудистого гемоглобинолиза – метгемальбуминемия и гемосидеринурия; часто развивается панцитопения, и причиной обращения к врачу могут быть кровоподтеки из-за тромбоцитопении. Изредка число лейкоцитов и тромбоцитов падает столь же сильно, сколь при тяжелой апластической анемии.
Нарушения пролиферации эпителиальных клеток ведкт к глосситу и заедам, или ангулярному стоматиту. Их можно выявить под микроскопом в слизистой щек, бронхов, мочевого пузыря и шейки матки.
У небольшой доли больных развивается гиперпигментация кожи, обусловленная избытком меланина. При авитаминозе В12 возможно поражение нервной системы различной тяжести: демиелинизация задних и боковых канатиков спинного мозга – фуникулярный миелоз, полинейропатия и нейропатия зрительного нерва. Появляется двусторонняя симметричная неврологическая симптоматика, обычно более выраженная на ногах и включающая покалывание, неустойчивость при ходьбе, падения в темноте, нарушение чувствительности и снижение мышечной силы. Реже наблюдаются нарушение зрения и психические расстройства.
Показано, что прием фолиевой кислоты до зачатия и на ранних сроках беременности снижает риск дефектов нервной трубки у плода: позвоночной расщелины, анэнцефалии и энцефалоцеле. Дефекты нервной трубки наблюдаются даже тогда, когда согласно лабораторным данным (нормальный уровень фолиевой кислоты в сыворотке или эритроцитах) у матери данного авитаминоза нет. При этом чем ниже сывороточная концентрация фолиевой кислоты или витамина В12 либо содержание фолиевой кислоты в эритроцитах, тем выше риск дефектов нервной трубки у плода. Обнаружена связь между дефектами нервной трубки и распространенной мутацией гена метилентетрагидрофолатредуктазы – заменой цитозина на тимин в положении 677 (см.2.7). Мутантный ген кодирует термолабильную форму фермента. Считается что профилактический прием фолиевой кислоты устраняет эти, а также другие, пока не установленные, нарушения обмена фолиевой кислоты и витамина В12. Мутация гена метилентетрагидрофолатредуктазы сопровождается повышением концентрации гомоцистеина в плазме беременной. Прием фолиевой кислоты или препаратов витамина В12 предотвращает гомоцистеинемию Каким образом нарушение метаболизма гомоцистеина приводит к дефектам нервной трубки, до сих пор не ясно.
Повышение концентрации гомоцистеина в плазме сопряжено также с повышенной частотой артериальных и венозных тромбозов. Причиной гомоцистеинемии могут быть дефицит фолиевой кислоты, авитаминозы В12 и В6, курение и злоупотребление алкоголем. У женщин в постменопаузе, не получающих заместительную гормональную терапию, и у мужчин концентрация гомоцистеина выше, чем у женщин детородного возраста и женщин в постменопаузе, получающих заместительную гормональную терапию. С возрастом гомоцистеинемия нарастает. Наследственное нарушение обмена метионина – гомоцистинурия – обусловлено дефектом одного из трех ферментов: цистатионинсинтетазы, метионинсинтазы или метилентетрагидрофолатредуктазы. При гомоцистинурии уже в детстве или в молодости развиваются тромбоэмболические заболевания.
Картина крови
Типичные для мегалобластной анемии показатели общего анализа крови представлены в табл.2.53. В мазке крови обнаруживают овальные макроциты, обломки эритроцитов (шизоциты), пойкилоциты и гиперсегментированные нейтрофилы – с ядрами, состоящими более чем из пяти сегментов (некоторые из этих клеток могут представлять собой макрополициты). Выраженность изменений картины крови соответствует тяжести заболевания. Вследствие экстрамедуллярного кроветворения (в печени и селезенке) у большинства больных в крови появляются мегалобласты(см.2.9). Внутри эритроцитов можно увидеть кольца Кебота – базофильные, богатые аргинином и содержащие негемовое железо образования (см.2.9, врезка). Если селезенки нет, например у перенесших гастроспленэктомию больных, или она атрофирована, как у 15% взрослых с целиакией, то в крови особенно выражены изменения, вызванные аспленией или функциональным аспленизмом (см. 2.10). В крайне тяжелых случаях вследствие усиленной фрагментации эритроцитов средний эритроцитарный объем может вернуться к норме. Приведем типичную гематологическую характеристику В12 и фолиеводефицитной анемии: мегалобластическая, макроцитарная, гиперхромная, диспластическая, с выраженным анизо- и пойкилоцитозом, в эритроцитах патологические включения в виде колец Кебота и телец Жолли. Сопровождается лейкопенией.
Картина костного мозга
При тяжелой мегабластной анемии значительно повышена клеточность костного мозга и наблюдается относительное преобладание ранних предшественников эритроцитов вследствие гибели поздних (см.2.11). Возможна инверсия лейкоэритробластического соотношения. В мегалобластах отмечается асинхронное созревание ядра и цитоплазмы: нежная сеть хроматина сопутствует нормально гемоглобинизированной цитоплазме. Кроме того, имеются признаки дизэритропоэза: избыток многоядерных клеток. межядерные хроматиновые мостики и тельца Говелла-Жолли. Присутствуют также гибнущие клетки (см.2.12).
Обнаруживают гигантские метамиелоциты неправильной формы (см.2.13). Мегекариоциты содержат гиперсегментированные ядра с нежной сетью хроматина. (см. 2.14).В легких случаях асинхронность созревания ядра и цитоплазмы определяется только в поздних предшественниках эритроцитов: это так называемые промежуточные мегалобластные изменения (см 2.15).При сочетании мегалобластной анемии с дефицитом железа развивается анемия, характеризующаяся эритроцитарным диморфизмом, то есть наличием двух популяций эритроцитов – хорошо гемоглобинизированных макроцитов и гипохромных микроцитов (см.2.16,А). Мегалобластные изменения в предшественниках эритроцитов могут быть незаметны,даже когда в костном мозге обнаруживают гигантские метамиелоциты (см.2.16,Б). В отсутствие дефицита железа обычно наблюдается избыточное образование гранул гемосидерина в эритрокариоцитах; в ряде случаев, прежде всего у больных, злоупотребляющих алкоголем, обнаруживают кольцевые сидеробласты, но с началом лечения они исчезают (см.2.17). Трепанобиопсия подтверждает скопление в костном мозге предшественников эритроцитов и повышенную митотическую активность (см.2.18). Примечательно, что у плода эритропоэз также протекает по мегалобластному типу (см.2.19).
Причины мегалобластных анемий
Дефицит витамина В12
Поскольку запасы витамина В12 в организме составляют 2-3 мг, а суточная потеря и, следовательно потребность в нем равны 1-2 мкг, дефицит развивается не раньше чем через 2-4 года с момента прекращения поступления витамина В12 с пищей или нарушения его всасывания. Авитаминоз В12 вследствие чрезмерных потерь или усиленного распада витамина В12 не описан. Закись азота (ингаляционный анестетик) способна быстро инактивировать витамин В12, превращая его из полностью восстановленной формы, кобаламина (I), в окисленные формы – кобаламин (II) и кобаламин (III). При длительной анестезии закисью азота развиваются мегалобластные изменения.
В отличие от фолиевой кислоты, которая содержится в большинстве продуктов, включая фрукты, овощи и крупы, витамин В12 присутствует только в продуктах животного происхождения. Строгое вегетарианство как причина авитаминоза В12 чаще всего встречается в Индии. Наибольшее количество фолиевой кислоты и витамина В12 содержится в печени – органе, запасающем питательные вещества.
Глубокий дефицит витамина В12, о котором судят по сывороточной концентрации этого витамина, не всегда сопровождается тяжелой анемией, но и в ее отсутствие может вызвать демиелинизацию задних и боковых канатиков спинного мозга, сопровождающуюся нейропатией. Это заболевание, известное как фуникулярный миелоз, чаще наблюдается у мужчин. Напротив, болезнь Аддисона – Бирмера – основная причина тяжелого авитаминоза В12 – чаще поражает женщин.
Болезнь Аддисона – Бирмера (известная также как пернициозная анемия) служит главной причиной авитаминоза В12 в западных странах. Она встречается повсеместно, но наиболее распространена в Северной Европе. Характерны ранняя седина, витилиго и заболевания щитовидной железы (см.2.20). Кроме того, болезнь Аддисона – Бирмера сочетается с другими органоспецифическими аутоиммунными заболеваниями, в частности с первичной надпочечниковой недостаточностью и гипопаратиреозом. У больных обнаруживают атрофический гастрит (см.2.21) с ахлоргидрией и циркулирующие аутоантитела: у 90% - к обкладочным клеткам желудка, у 50% - к внутреннему фактору Касла. Рак желудка при болезни Аддисона – Бирмера развивается в 2-3 раза чаще, чем среди населения в целом. Авитаминоз В12 развивается также при избыточном росте бактерий в тонкой кищке, например вследствие дивертикулеза тощей кишки, при болезни Крона, после резекции повздошной кишки.

