




1.1. Особенности термодинамического поведения
реального газа
В паротурбинных и холодильных установках рабочие тела следует рассматривать как реальные газы. Между свойствами идеального и реального газов имеются существенные различия, обусловленные влиянием сил межмолекулярного взаимодействия и наличием собственного объёма молекул реального газа.
Для анализа поведения реального газа рассмотрим на диаграмме р,v процесс изотермического сжатия реального газа (рис. 1.1). При температурах выше критической (см. табл. 1.1), а также при больших значениях удельного объёма изотермы реального газа близки к равнобочным гиперболам, как у идеального газа. При изотермическом сжатии газа при Т<Ткр его давление монотонно возрастает, пока не достигнет определённого значения, называемого давлением насыщенного пара ps, находящегося в равновесии с жидкостью (процесс 1–2). При дальнейшем сжатии давление пара не изменяется, но он конденсируется –– переходит в жидкое состояние (процесс 2–3). Когда весь пар сконденсируется, начнётся изотермическое сжатие жидкости; здесь давление резко возрастает при незначительном уменьшении удельного объёма (процесс 3–4).
Таблица 1.1. Критические параметры некоторых веществ
| Веще- ство | Параметры | Веще- ство | Параметры | ||||
| Т кр, К | р кр, МПа | ρкр, кг/м3 | Т кр, К | р кр, МПа | ρкр, кг/м3 | ||
| H2 | 33,2 | 1,3 | СО2 | 304,2 | 7,38 | ||
| N2 | 126,2 | 3,40 | NH3 | 405,5 | 11,28 | ||
| O2 | 154,6 | 5,04 | H2O | 647,3 | 22,11 |
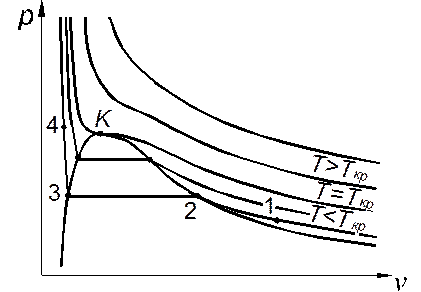 Рис. 1.1. Изотермы реального газа на диаграмме p,v.
Рис. 1.1. Изотермы реального газа на диаграмме p,v.
|
Точки 2 и 3 соответствуют характерным состояниям пара ижидкости при фазовом равновесии: 2 — насыщенному пару, 3 — насыщенной (кипящей) жидкости. Участок 1–2 соответствует паровой ветви изотермы, 3–4 — жидкостной, а участок конденсации 2–3 –– состояниям влажного пара, то есть равновесной смеси паровой и жидкой фаз; на этом участке изотерма одновременно является изобарой.
Значение удельного объёма насыщенного пара v" на большинстве докритических изотерм гораздо больше значения удельного объёма насыщенной жидкости v'. С ростом температуры разность v"– v' уменьшается и становится равной нулю при критической температуре в критической точке К, в которой исчезают различия между свойствами кипящей жидкости и насыщенного пара. Критическая изотерма Т = Т кр на диаграмме p,v имеет в критической точке горизонтальную касательную и точку перегиба, что математически выражается критическими условиями

| (1.1) |
Кривая 2–К–3 на термодинамических диаграммах p,v и T,v, отделяющая область двухфазного состояния (влажного пара) от однофазной, называется пограничной кривой. Она состоит из двух ветвей: пограничной кривой жидкости К–3 на диаграмме p,v и К-2 на диаграмме T,v и пограничной кривой пара К–2 на диаграмме p,v и К-3 на диаграмме T,v, которые плавно сопрягаются в критической точке.
При изобарном подводе теплоты к жидкости, взятой при низкой температуре Т 1, и давлении, меньшем критического, температура жидкости повышается до температуры насыщения Тs=T 2, то есть до температуры фазового равновесия жидкости и пара (процесс 1–2 на диаграмме Т,v, рис. 1.2). Удельный объём жидкости при её нагреве от Т 1до Т 2увеличивается незначительно. При дальнейшем подводе теплоты при постоянном давлении происходит парообразование — фазовый переход вещества из жидкого в парообразное состояние (процесс 2–3). Температура в процессе парообразования остается постоянной и равной Тs. После превращения всей жидкости в пар подвод теплоты приводит к росту температуры пара (к его перегреву).
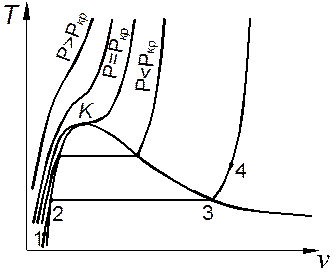 Рис. 1.2. Изобары реального газа на диаграмме Т,v.
Рис. 1.2. Изобары реального газа на диаграмме Т,v.
|
Итак, на докритической изобаре можно выделить три характерных участка: 1–2 — нагрев жидкости до температуры насыщения, 2–3 —парообразование (кипение), 3–4 — перегрев пара. Противоположный процесс — изобарный отвод теплоты –– приводит к охлаждению пара и его конденсации, а при дальнейшем отводе теплоты жидкость охлаждается и затем затвердевает.
С ростом давления увеличивается температура насыщения и уменьшается разность v"– v'. При критическом давлении v' = v" = vкр. Изобара p = p кр в критической точке К имеет горизонтальную касательную и точку перегиба.
Из диаграмм р,v и Т,v видно, что каждому значению давления насыщенного пара ps соответствует определённое значение температуры Ts ё. Кривая ps=f(Ts) на фазовой диа-
грамме р,Т (рис.1.3), соответствующая равновесному сосуществованию жидкости и пара, называется кривой парообразования. Максимальные значения р и Т на данной кривой –– это параметры критической точки К, а минимальные — параметры тройной точки L, в которой находятся в равновесии три фазы: паровая, жидкая и твердая.
 Рис. 1.3. Фазовая диаграмма реального газа.
Рис. 1.3. Фазовая диаграмма реального газа.
|
На диаграмме р,Т показаны также изохоры, каждая из которых представляет собой линию малой кривизны, исходящую из точки на кривой парообразования. Изохоры пара, которым соответствуют значения v>v кp, расположены правее кривой парообразования 1, а изохоры жидкости при v<v кp — левее. Изохора v=v крисходит из критической точки и имеет в ней общую касательную с кривой парообразования, что выражается правилом Планка-Гиббса
 . .
| (1.2) |
Такое термодинамическое поведение реального газа свойственно всем индивидуальным веществам в характерных для каждого из них интервалах изменения параметров. Оно гораздо сложнее поведения идеального газа.
На полной фазовой диаграмме (рис. 1.4) показаны три фазы, разделённые линиями фазового равновесия 0–L, L–К и L–N, сходящимися в тройной точке L. Определив по параметрам Т и р положение точки на диаграмме, можно установить состояние вещества. Положение точки на линии фазового равновесия свидетельствует о том, что сосуществуют две фазы. Линия О–L соответствует равновесию твердой и газообразной фаз, линии L–N и L–К –– равновесию твердой–жидкой и жидкой–газообразной фаз. В критической точке К заканчивается линия равновесия жидкость–пар.
 Рис. 1.4. Фазовые диаграммы для веществ, плотность которых при плавлении уменьшается (а) либо увеличивается (б).
Рис. 1.4. Фазовые диаграммы для веществ, плотность которых при плавлении уменьшается (а) либо увеличивается (б).
|
Для большинства веществ наклон линии равновесия твердое тело–жидкость положителен, как показано на рис. 1.4 а, что обусловлено уменьшением плотности вещества при плавлении. В этом случае повышение давления жидкости при T = const приводит к затвердеванию вещества (линия 1–2 на рис 1.4 а). Однако у некоторых веществ плотность при плавлении возрастает, у них наклон линии равновесия твердое тело–жидкость отрицателен (см. рис. 1.4 6). Пример такого вещества –– вода; лёд при изотермическом сжатии плавится (линия 1–2 на рис. 1.4 6).
На диаграмме р,Т (рис. 1.4) изобарный процесс 3–4 соответствует сначала нагреву твердого тела, затем его плавлению при температуре Т пл и давлении р пл и, наконец, нагреву жидкости. Линия 3–4 пересекает кривую равновесия твёрдое тело–жидкость. При продолжении этого процесса (линия 4–4')происходит нагрев жидкости, парообразование при температуре Т пи давлении р п, а затем перегрев пара. Линия 4–4' пересекает кривую равновесия жидкость–газ. Если давление меньше давления в тройной точке, твердое тело при изобарном нагреве переходит в газообразное состояние, минуя жидкую фазу. Это процесс сублимации (возгонки) 5–6.
У аморфных веществ, не имеющих в твёрдом состоянии кристаллической структуры (например, у воска), переход из твёрдого состояния в жидкое при постоянном давлении происходит при переменной температуре.
1.2. Уравнения состояния и таблицы термодинамических свойств реального газа
Применение методов термодинамики для решения технических задач невозможно без знания термодинамических свойств рабочих веществ. Экспериментальное определение свойств реальных газов –– трудоёмкий процесс, и не все необходимые для расчётов свойства могут быть получены в эксперименте (например, энтропия). В то же время по термическому уравнению состояния p=f (T, v)можно рассчитать не только термические (р, v, T),но и калорические свойства (h, s, cv, cp), так как на основании первого и второго законов термодинамики получены термодинамические соотношения, связывающие эти свойства.
В связи со сложностью термодинамического поведения реальных газов уравнение состояния реального газа имеет сложную структуру. Среди уравнений состояния одно из наиболее простых –– уравнение Ван-дер-Ваальса
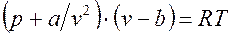 , ,
| (1.3) |
где а и b — постоянные, учитывающие соответственно парное взаимодействие молекул и их собственный объём.
Это уравнение качественно правильно описывает почти все особенности поведения реального газа. Однако оно даёт большие погрешности вблизи пограничной кривой, в критической области и в области жидкой фазы.
М.П. Вукалович и И.И. Новиков предложили более сложное уравнение состояния, учитывающее объединение молекул в комплексы из двух, трёх и бὀльшего числа молекул. Это явление влияет на свойства вещества в области высоких плотностей. Такое уравнение, использовавшееся для расчета свойств воды и водяного пара, имеет вид
 , ,
| (1.4) |
где А 1(Т), А 2(Т), ... — функции температуры,
а и b — постоянные, аналогичные постоянным в (1.3).
Общее уравнение состояния реального газа, теоретически обоснованное Дж. Майером и Н.Н. Боголюбовым методами статистической физики, записывается в форме

| (1.5) |
Функции температуры В (Т), С (Т), ... — вириальные коэффициенты, учитывающие парные, тройные и более сложные взаимодействия молекул. При v →∞ уравнение (1.5) переходит в уравнение состояния идеального газа.
На практике в уравнении (1.5) ряд заменяют полиномом. Функции В (Т), С (Т),... также представляют в виде по-
линомов от 1/ Т, их коэффициенты определяют по опытным данным о термодинамических свойствах вещества.
Уравнение состояния содержит в компактной форме информацию о свойствах вещества и может использоваться для расчётов процессов и циклов с помощью ЭВМ. Однако во многих инженерных расчётах применяются не эти сложные уравнения, а составленные с их помощью таблицы термодинамических свойств и диаграммы состояния. Для воды и водяного пара М.П. Вукалович, С.Л. Ривкин и А.А. Александров рассчитали таблицы свойств, которые широко используются при тепловых расчётах.
На кафедре термодинамики ОИИМФ в 1960–1995 гг. составлены уравнения состояния и рассчитаны таблицы термодинамических свойств воздуха и его компонентов, метана, этана, пропана и других технически важных газов. Ряд таблиц был утверждён Госстандартом СССР в качестве таблиц стандартных справочных данных, обязательных к применению во всех отраслях народного хозяйства.
Для расчёта калорических свойств вещества с помощью уравнения состояния используются термодинамические соотношения, которые получаются из объединённого уравнения I и II законов термодинамики:
 . .
| (1.6) |
Для вывода этих соотношений выразим в уравнении (1.6) полные дифференциалы ds и du через частные производные
 . .
| (1.7) |
Для частного случая изохорного процесса (dv = 0), учитывая известное равенство (∂ u/∂T) v = cv, из уравнения (1.7) получим
 . .
| (1.8) |
Для изотермического процесса (dT = 0) из (1.7) следует
 . .
| (1.9) |
Продифференцировав (1.9) по Т при v = const, найдём
 . .
| (1.10) |
Для смешанной производной порядок дифференцирования безразличен, и с учетом равенства (1.8) можно записать
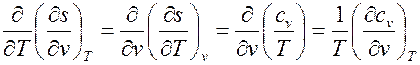 , ,
| (1.11) |
 . .
| (1.12) |
Подставив полученные выражения в (1.10) и исключив одинаковыечлены в левой и правой частях, получим
 . .
| (1.13) |
Это выражение представляет собой дифференциальное термодинамическое соотношение, связывающее частные производные термических и калорических величин.
Подставив выражение (1.13) для производной (∂ s /∂ v) T в равенство (1.9), можно записать уравнение для расчета частной производной (∂ u /∂ v) T через термические параметры
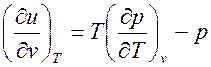 , ,
| (1.14) |
где правую часть можно вычислить из уравнения состояния.
Дифференцируя соотношение (1.13) по Т и учитывая (1.8), получим
 , ,
| (1.15) |
или окончательно

| (1.16) |
Аналогичным путём могут быть получены и другие термодинамические соотношения, связывающие в дифференциальной форме термические и калорические свойства веществ.
Использование термодинамических соотношений для определения калорических свойств можно проиллюстрировать на примере расчёта внутренней энергии на основании соотношения (1.14), интегрируя которое получим

| (1.17) |
При расчётах внутренней энергии в (1.17) следует подставить выражения функций р и (∂ p /∂ T) v, полученные из уравнения состояния p = f (T, v). После подстановки интегрируем при постоянной температуре, в результате чего на каждой изотерме появляется своя постоянная интегрирования φ. Совокупность этих постоянных образует неизвестную функцию φ(T).
Для определения функции φ(T) рассмотрим предельный случай p →0, когда уравнение состояния приобретает вид p = RT / v. Тогда подынтегральное выражение в (1.17) обращается в нуль и видно, что функция φ(T) является внутренней энергией идеального газа. Окончательно получим
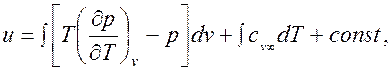
| (1.18) |
где постоянная интегрирования определяется началом отсчёта внутренней энергии идеального газа. Аналогично из соотношений (1.13) и (1.16) можно получить выражения для расчёта энтропии и изохорной теплоемкости реального газа

| (1.19) |

| (1.20) |
Взяв вторую форму основного уравнения термодинамики, представленную через переменные Т и р, запишем объединённое уравнение обоих законов термодинамики в виде

| (1.21) |
На основании уравнения (1.21) можно получить дифференциальные термодинамические соотношения при переменных Т и р. Для их использования необходимо располагать уравнением состояния в форме v = F (T, p).
Термодинамические соотношения имеют важное прикладное и теоретическое значение. Они позволяют на основании термического уравнения состояния вещества рассчитывать его калорические свойства, которые экспериментально определяются сложнее и менее точно, чем термические свойства. Это сокращает объём трудоёмких экспериментальных исследований калорических свойств и в тоже время позволяет составлять подробные таблицы термодинамических свойств реальных газов.
Хорошее согласование рассчитанных значений калорических свойств с опытными данными свидетельствует о точности и взаимной согласованности использованных данных о термических и калорических свойствах вещества, а также о строгости термодинамических соотношений и справедливости I-го и II-го законов термодинамики, на основании которых они получены. Последнее обстоятельство определяет теоретическое значение этих соотношений.
Таблицы свойств пара и жидкости в состоянии насыщения являются однопараметрическими, поскольку свойства на пограничной кривой определяются одним параметром состояния. В качестве независимой переменной выбирают либо температуру, либо давление.
Таблицы свойств однофазной области (в состоянии жидкости либо пара) составляют в зависимости от двух переменных — давления и температуры  . Иногда в верхней части таблицы для каждой изобары повторяют данные о свойствах насыщенных пара и жидкости, что позволяет решать ряд задач, не прибегая к таблицам для состояния насыщения. Жирная горизонтальная линия в таблице на изобарах при давлениях ниже критического разделяет данные о свойствах жидкости и перегретого пара.
. Иногда в верхней части таблицы для каждой изобары повторяют данные о свойствах насыщенных пара и жидкости, что позволяет решать ряд задач, не прибегая к таблицам для состояния насыщения. Жирная горизонтальная линия в таблице на изобарах при давлениях ниже критического разделяет данные о свойствах жидкости и перегретого пара.
Таблицы составляют с такими шагами по температуре и давлению, которые позволяют применять линейную интерполяцию при промежуточных значениях Т и р.
Используя данные для состояния насыщения, можно определять свойства влажного пара, если известна степень сухости, то есть массовая доля насыщенного пара во влажном паре. Тогда 1 кг влажного пара как смеси содержит (1- х)кг насыщенной жидкости и х кг насыщенного пара. Свойства этих фаз обозначают одним и двумя штрихами. Определив по таблицам для заданной температуры либо давления значения v', v", h', h", s', s",можно рассчитать свойства влажного пара по правилу аддитивности:
  , ,
| (1.22) |
 , ,
| (1.23) |
 . .
| (1.24) |
При малых давлениях (температурах) v" намного больше v' и значения vx можно рассчитывать по формуле vx≈v"x.
Разность h" – h' = r — это количество теплоты, подведенной в процессе фазового перехода вещества из состояния кипящей жидкости в состояние насыщенного пара. Величина r называется теплотой парообразования; она уменьшается с ростом давления насыщенного пара и равна нулю при р = ркр. В таблицах обычно даны значения r,их удобно использовать при расчёте по формуле (1.23).
1.3. Термодинамические диаграммы реального газа
Для облегчения расчётов процессов с реальным газом используют термодинамические диаграммы, построенные по табличным данным. Достоинство диаграмм по сравнению с таблицами –– наглядность, позволяющая проводить качественный анализ процессов и циклов, а недостаток — меньшая точность. На практике большую роль играет изоэнтропный процесс, поэтому широкое распространение получили энтропийные диаграммы Т, s и h, s,на которых этот процесс изображается вертикальной прямой линией.
На рис. 1.5 приведена диаграмма Т, s для реального газа. На диаграмме нанесена сетка изобар, конфигурация которых аналогична конфигурации этих кривых на диаграмме Т, v (см. рис 1.2). На докритических изобарах левая ветвь кривой характеризует состояния жидкости, правая — перегретого пара, а горизонтальный участок изобары ––
 Рис. 1.5. Диаграмма T, s для реального газа.
Рис. 1.5. Диаграмма T, s для реального газа.
|
состояния влажного пара. Двухфазная область отделена от однофазной пограничной кривой, максимум которой соответствует критической точке К. Левая и правая ветви этой кривой на диаграмме Т, s примерно симметричны.
Критическая изобара касается пограничной кривой в критической точке, где имеет горизонтальную касательную и точку перегиба. Сверхкритические изобары представляют собой монотонные кривые, имеющие перегибы. В области жидкости изобары близки к пограничной кривой. По мере роста температуры и понижения давления конфигурация изобар в однофазной области приближается к конфигурации изобар идеального газа в этихкоординатах.
На диаграмме Т, s также наносят изохоры, которые круче изобар (как и для идеального газа). В двухфазной области изохоры — наклонные кривые малой кривизны, обращенные выпуклостью вверх. При переходе из однофазной области в двухфазную изохоры имеют излом, которому соответствует скачок изохорной теплоёмкости.
На диаграмме Т, s часто наносят линии h = const (изоэнтальпы). Они располагаются тем выше, чем больше значение энтальпии. С понижением давления эти линии стремятся к горизонталям, так как свойства пара приближаются к свойствам идеального газа. Некоторые изоэнтальпы имеют максимум, соответствующий точке на кривой инверсии. Эта кривая отделяет область параметров, в которой дроссельный эффект α h = (дТ/др) h положителен, от области, где α h <0. Положение кривой инверсии и упомянутых областей показано на диаграмме р,Т (рис. 1.6), а особенности процесса дросселирования рассматриваются в § 2.5.
 Рис. 1.6. Кривая инверсии (1) и кривая парообразования (2): α h >0 – область понижения температуры газа при дросселировании, α h <0 – область повышения температуры газа.
Рис. 1.6. Кривая инверсии (1) и кривая парообразования (2): α h >0 – область понижения температуры газа при дросселировании, α h <0 – область повышения температуры газа.
|
Пользуясь формулой (1.24) для энтропии влажного пара, наносят в двухфазной области диаграммы T, s линии x =const (на рис. 1.5 они не нанесены). Предельным значениям х= 0 и х= 1соответствуют пограничные кривые жидкости и пара. Все линии х= сonst сходятся в критической точке.
На диаграмме Т, s площадь под кривой процесса изображает теплоту процесса, поэтому диаграмма называется тепловой. Площадь под участком изобары 1–2 соответствует теплоте, идущей на нагрев жидкости от температуры Т 1 до температуры насыщения Тs, площадь под участком 2–3 — теплоте парообразования, а площадь под участком 3–4 — теплоте перегрева пара от температуры насыщения до Т 4.
Диаграмма Т, s с сетками изобар, изохор и изоэнтальп называется полной, поскольку с её помощью можно выполнять расчеты, не привлекая уравнение состояния либо таблицы термодинамических свойств. Тем не менее, желательно помимо диаграммы использовать таблицы, так как значения удельного объёма и энтальпии определяются по ней с большой погрешностью, особенно в области жидкости.
Тепловая диаграмма удобна для анализа циклов, но для инженерных расчетов чаще применяют диаграмму h, s (рис. 1.7),с помощью которой значения энтальпии определяются проще и точнее. На диаграмме h, s критическая точка расположена не в максимуме пограничной кривой, а левее, так как энтальпия насыщенного пара достигает максимума при докритическом давлении (для водяного пара при р =3 МПа), а при дальнейшем росте давления убывает.
 Рис. 1.7. Диаграмма h, s для реального газа
Рис. 1.7. Диаграмма h, s для реального газа
|
На диаграмме h, s нанесены изобары, изохоры, а также изотермы в области перегретого пара и линии x = const в области влажного пара, в которой изотермы совпадают с изобарами. Для влажного пара изобары — прямые линии, так как их тангенс угла наклона (∂ h /∂ s) p = T в этой области равен температуре насыщения Ts то есть постоянен для изобары.
Чем выше давление насыщения (а следовательно и температура насыщения), тем круче изобара в двухфазной области. Этим обусловлен веерообразный ход изобар в двухфазной области на диаграмме h, s. В отличие от диа грамм T, v и T, s, на диаграмме h, s изобары плавно переходят из двухфазной области в однофазную, так как в точке перехода для обоих участков изобары (∂ h /∂ s) p = Ts.
Из соотношения (∂ h /∂ s) p = T очевидно, что кривизна изобар на диаграмме h,s всегда положительна. Следовательно, все изобары, в том числе и сверхкритические, не имеют перегибов. Изохоры, как и на диаграмме T, s, имеют бὀльшую крутизну, чем изобары.Кривые х= сonst на диаграмме h, s рассчитывают и строят так же, как на диаграмме T, s.
Изотермы на диаграмме h, s с ростом энтропии (то есть с понижением давления) стремятся к горизонтальным прямым, соответствующим изотермам идеального газа. На ряде изотерм имеются минимумы, геометрическое место которых на этой диаграмме образует кривую инверсии.
При малых значениях x, а также при высоких давлениях и низких температурах изобары сближаются и примыкают к пограничной кривой жидкости. Поэтому диаграмма h, s обычно охватывает не всю двухфазную область, а лишь часть её при значениях x> 0,65.
На диаграмме h, s теплота изобарного процесса и техническая работа изоэнтропного процесса, равные разности значений энтальпии, изображаются отрезками (а не площадями, как на диаграмме Т, s). Это упрощает расчёты и обусловило широкое применение диаграммы h, s на практике.
При помощи диаграмм Т, s и h, s можно определять свойства перегретого пара и жидкости, если заданы значения давления и температуры, и свойства влажного пара, если известны значения степени сухости и давления (либо температуры). С помощью диаграммы h, s можно найти температуру насыщения пара по его давлению, определив, какой изотерме соответствует точка пересечения данной изобары с пограничной кривой. Хотя на диаграмме h, s пограничная кривая жидкости не представлена, для низких и умеренных давлений можно с приемлемой точностью определить значение энтальпии воды в состоянии насыщения, необходимое при расчётах циклов паротурбинных установок, по формуле

| (1.25) |
где ts,°С— температура насыщения при данном давлении,
размерность значения энтальпии — кДж/кг.
Коэффициент 4,1868 в формуле (1.25) представляет собой изобарную теплоёмкость воды в кДж/(кг.К), которую при невысоких давлениях можно считать постоянной. При давлении насыщения менее 3 МПа погрешность значений энтальпии, рассчитанных по (1.25), не превышает 3 %.
При расчётах циклов парокомпрессорных холодильных установок применяют специальную диаграмму h,ln p, а при расчетах процессов во влажном воздухе –– диаграмму H, d (энтальпия–влагосодержание). Диаграмма h,ln p рассматривается в курсе холодильных установок, а диаграмма H, d описана в последнем разделе данного пособия.

