




Гаргоилизм или болезнь Гурлер - Пфаундлера - Хантера, мукополисахаридоз, липохондродистрофия, множественный дизостоз) - группа заболеваний, обусловленных наследствен мои патологией соединительной ткани, различающихся по характеру обменных нарушений, но имеющих большое клиническое сходство. Характеризуются одновременным поражением центральной нервной системы, органов зрения, опорно- двигательного аппарата и внутренних органов. Заболевание описано Thomson, Huntef, Hurler' Pfaundler, заболевание изучалось Л. О. Бадаляном, Е. И. Гусевьм. Название «гаргоилизм» дано заболеванию в связи с некоторым внешним сходством больных со скульптурными изображениями химер (gargoiles).
Заболевание передается по аутосомно- рецессивномутипу наследования или рецессивному, сцепленному с Х- хромосомой (формаHunter). Мальчики заболевают в 2 раза чаще, чем девочки. Для возникновения заболевания имеет значение кровное родство родителей. В основе заболевания лежат генетически обусловленные нарушения обмена кислых мукополисахаридов, сложных белково- полисахаридных образований, которые играют важную роль в структуре и функционировании соединительной ткани. У больных обнаруживается избыточное выделение их с мочой и накопление в соединительной ткани и внутренних органах. Так как в различных органах и тканях преобладают различные типы мукополисахаридов, нарушение их обмена, особенно кислых гликозамингликонов, приводит к различным симптомокомплексам.
| 
| 
|
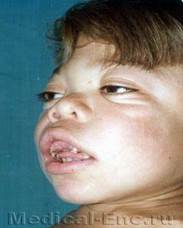
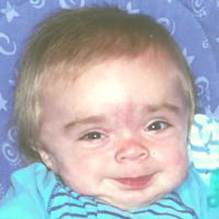
Рис. 1.Гаргоилизм
При патологоанатомическом исследовании обнаруживаются изменения во многих органах и системах. Высокомолекулярные липоидно- полисахаридные вещества откладываются в виде мелкозернистой массы в нервных клетках мозга периферических ганглиях и сетчатке глаз. Происходит значительное изменение мозга, возникают гидроцефалия, утолщение твердой мозговой оболочки. В костной системе обнаруживаются нарушения энхондрального и перихондрального окостенения, разрастание кровеносных сосудов в хряще накопление в его клетках гликозамингликонов и липидов. Эти же вещества откладываются в паренхиматозных клетках печени ретикулярных клетках селезенки, эпителии извитых канальцев почек, а также в сердце, аорте и других органах. В основном веществе соединительной ткани обнаруживается накопление только гликозамингликонов. В нем отмечается утолщение коллагеновых волокон и дезорганизация основного вещества.
Типовые клинические признаки заболевания проявляются в первые месяцы жизни ребенка. Характерен внешний вид больных. Постепенно нарастают явления акромегалии, становятся грубыми черты лица. Череп увеличивается в размерах, отмечаются дисплазии: лоб нависает над лицом нос широкий с запавшей переносицей, выражен гипертелоризм и экзофтальм, деформированные ушные раковины расположены низко, губы сплюснуты, неправильный рост зубов, высокое небо, язык увеличен в размерах (мегалоглоссия). Постепенно эти диспластические признаки становятся более выраженными. Шея, туловище и конечности больных укорочены. Грудная клетка деформирована, реберные дуги развернуты кнаружи, позвоночник искривлен. Ладонь широкая, пальцы короткие, толстые. Отмечается тугоподвижность в суставах. При рентгенологическом исследовании выявляется уплотнение костей черепа, расхождение швов и истончение их краев, уплощение турецкого седла. В трубчатых костях обнаруживается утолщение эпифизов, нарушение энхондрального и перихондрального окостенения.
Со стороны внутренних органов обнаруживаются пороки сердца, диффузные изменения миокарда, явления гепатоспленомегалии, увеличение живота, пупочные и паховые грыжи. Слух и зрение постепенно снижаются. При офтальмологическом исследовании нередко выявляются помутнение роговицы, мегакорнеа, катаракты, глаукома, застойные явления на глазном дне, атрофия дисков зрительных нервов. Со стороны центральной нервной системы отмечаются резкая диффузная мышечная гипотония, двигательная заторможенность, вялость. Психическое развитие детей резко задерживается. Психические процессы отличаются чрезвычайной вялостью и инертностью. Интеллектуальный дефект прогредиентно нарастает и чаще достигает степени идиотии. Степень выраженности, частота и сочетание различных клинических признаков при мукополисахаридозах не всегда одинаковы. По типу наследования, выраженности клинических проявлений, степени прогредиентности и особенностям биохимических нарушений различают шесть типов (синдромов) заболевания.
Диагностика гаргоилизма основывается на клинических, генеалогических и биохимических данных. Биохимическое обследование включает исследование мочи больных на кислые мукополисахариды (реакция Барри с толуидиновым синим, реакция преципитации кислых мукополисахаридов с альбумином - реакция Дорфмана). Отдельные фракции мукополисахаридов исследуются с помощью методов электрофореза на бумаге и хроматографии.
Специфическая патогенетическая терапия гаргоилизма не разработана. Рекомендуется назначение высоких доз АКТГ для угнетения синтеза кислых мукополисахаридов. В связи с недостаточностью функции щитовидной железы целесообразно назначение тиреоидина. Имеется указание на положительный терапевтический эффект применения высоких доз витамина А и рентгеновского облучения гипофиза.

