




| Исходное основание | Основание, занявшее место исходного | Тип замены |
| Пурин | Другой пурин | Простая замена (транзиция) |
| Пиримидин | Другой пиримидин | То же |
| Пурин | Любой пиримидин | Перекрестная замена (трансверсия) |
| Пиримидин | Любой пурин | Перекрестная замена (трансверсия) |
Спонтанные замены азотистых оснований происходят очень редко. Например, в соответствии с существующими расчетами у человека за год случается около 10—20 спонтанных замен оснований, причем одна замена может быть повторена на каждые 10 000 генов лишь 50 раз на протяжении времени в 1 млн лет. Можно полагать, что такая чрезвычайно низкая частота замен оснований в ДНК присуща как животным (млекопитающим), так и растениям. Спонтанные замены азотистых оснований возникают в ДНК в результате «ошибок», совершаемых ДНК-полимеразой и сопровождающихся неправильным спариванием оснований. Одно из объяснений этой «ошибочности» было дано Д. Уотсоном и Ф. Криком еще в 1953 г. и оно сводится к признанию в ошибочном спаривании роли тауто-мерных форм (структур, в которых протон перешел на место, противоположное обычной водородной связи) естественных оснований. Следовательно, структурные основы для мутаций в виде замен оснований обеспечивают таутомеры естественных оснований.
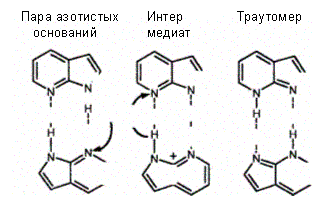 Транзиции индуцируются азотистой кислотой, которая вызывает окислительное дезаминирование аденина, цитозина и гуанина, содержащих свободные аминогруппы, в гипоксантин, урацил и ксантин соответственно. Из-за того, что дезаминирование сопровождается переходом аминооснования в кетонооснование, гипоксантин, например, подобно гуанину, будет спариваться с цитози-ном, т. е. в результате дезаминирования аденина в гипоксантин пара А-Т перейдет в пару Г-Ц. В случае дезаминирования цитозина в урацил пара Г-Ц перейдет в пару А-Т. Транзиции индуцируются также алкилирующими соединениями. Например, этилметан-сульфонат алкилирует гуанин и освобождает от него ДНК без нарушения ее сахарофосфатного каркаса. Следовательно, гуанин может быть заменен любым основанием, и это ведет не только к транзициям, но и к трансверсиям.
Транзиции индуцируются азотистой кислотой, которая вызывает окислительное дезаминирование аденина, цитозина и гуанина, содержащих свободные аминогруппы, в гипоксантин, урацил и ксантин соответственно. Из-за того, что дезаминирование сопровождается переходом аминооснования в кетонооснование, гипоксантин, например, подобно гуанину, будет спариваться с цитози-ном, т. е. в результате дезаминирования аденина в гипоксантин пара А-Т перейдет в пару Г-Ц. В случае дезаминирования цитозина в урацил пара Г-Ц перейдет в пару А-Т. Транзиции индуцируются также алкилирующими соединениями. Например, этилметан-сульфонат алкилирует гуанин и освобождает от него ДНК без нарушения ее сахарофосфатного каркаса. Следовательно, гуанин может быть заменен любым основанием, и это ведет не только к транзициям, но и к трансверсиям.
Транзиции часто вызываются мутагенами, действующими на ДНК только в состоянии репликации, например, 5-бромурацилом, который является аналогом тимина и способен включаться в ДНК посредством замещения тимина. Наряду с нормальной способностью 5-бромурапила спариваться с аденином иногда возникает состояние, когда он действует не как тимин, а как цитозин, что обеспечивает формирование водородных связей его не с аденином, а с гуанином. Эти «ошибки» спаривания происходят либо при включении 5-бро-мурацила в ДНК («ошибки» включения), либо при репликации ДНК после его включения («ошибки» репликации). Следовательно, время «ошибок» определяет характер транзиции. «Ошибки» спаривания, индуцируемые 5-бромурацилом, ведут к транзициям от пары Г-Ц к паре А-Т, и наоборот (от А-Т к Г-П). Подобные транзиции индуцируются также 2-аминопурином.
Замены оснований приводят к изменениям смысла кодонов, вследствие чего они приобретают способность кодировать другую аминокислоту (миссенс-мутации). Например, замена в триплете ГУА, содержащемся в гене р-гемоглобина, урацила на аденин (трансверсия) сопровождается тем, что в цепи р-гемоглобина вместо валина оказывается глутаминовая кислота. Это ведет к превращению гемоглобина в новый вариант мутантного гемоглобина (например, типа Бристоль). В результате замен оснований возникают также нонсенс-мутации, когда на измененных кодонах обрывается чтение информации гена (как правило, такими кодонами являются триплеты УАГ, УАА и УГА). Одновременно в результате замен образуются кодоны, сохраняющие исходный смысл.
Делеции и включения одного или нескольких азотистых оснований в нуклеотидных последовательностях ДНК могут быть ошибками репликации ДНК или индуцироваться акридиновыми красителями. Такие изменения называют мутациями сдвига рамки, ибо они приводят к сдвигу «рамки чтения» кода гена. Включаясь между соседними основаниями, акридин оранжевый заставляет их «раздвигаться» на расстояние в 0,6-0,8 нм.
Если акридин оранжевый присутствует в полинуклеотидной цепи-шаблоне, то результатом будет добавление основания в новую цепь в процессе репликации ДНК. Если же акридин оранжевый присутствует в клетке во время репликации ДНК, то он может включаться в новую цепь вместо основания, имитируя парное (противоположное) основание в цепи-шаблоне, и затем выйти. Это приводит к тому, что вновь реплицированной цепи будет недоставать основания, т. е. она будет реплицирована с делецией по основанию- Делеции могут затрагивать несколько оснований. Например, описаны делеции 15 оснований, которые сопровождались утратой в белке 5 аминокислот.
Дупликации (добавление) 1—2 оснований могут приводить также к мутациям со сдвигом «рамки считывания» кода. Если дупли-кация происходит внутри гена, то «рамка считывания» нарушается на большом протяжении.
Делеции и дупликации азотистых оснований представляют собой молекулярный механизм и мутации митохондриальной ДНК человека. Установлено, что из мтДНК человека могут быть делегированы сегменты длиной до 5000 пар оснований.
Особую форму молекулярных механизмов генных мутаций представляют повторы триплетов азотистых оснований. Наличие в молекулах ДНК повторов триплетов оснований сопровождается нарушениями нормального цикла репликации ДНК, с одной стороны, и аномальным синтезом белка (из-за повторов аминокислоты, кодируемой повторяющимся триплетом), с другой стороны. Например, мутации гена, контролирующего белок хантингтан, недостаток которого у человека сопровождается болезнью Хантингтона, заключаются в резком увеличении повторов триплета ЦАГ.
Репарация повреждений ДНК
Мутагенные и летальные эффекты мутагенов сопровождаются структурными повреждениями, которые они вызывают в молекулах ДНК. Например, в геноме человека непрерывно происходят случайные изменения (повреждения), но сохраняются лишь отдельные из них. Причем очень редко. Так из 1000 замен азотистых оснований лишь одна приводит к мутациям. Причина заключается в том, что эти повреждения часто подвержены восстановлению. Процесс реконструкции повреждений ДНК называют восстановлением или репарацией ДНК.
Характер и механизмы исправления повреждений наиболее полно изучены в случае повреждений, индуцированных УФ-излучени-ем. Клетки реагируют на УФ-излучение тем, что в их ДНК образуются повреждения, главные из которых представляют собой фотохимические изменения в пиримидиновых основаниях, переходящие в пиримидиновые димеры, в частности в тиминовые. Последние образуются за счет ковалентного связывания соседних тиминовых оснований в одной и той же цепи молекулы посредством присоединения углерода одного тимина к углероду другого тимина. Помимо тиминовых димеров в ДНК облученных клеток происходит формирование также цитозин-тиминовых и цитозин-цитозиновых димеров, однако частота их является меньшей. Димеризация фланкирующих оснований в гене сопровождается ингибированием транскрипции и репликации ДНК. Она ведет также к мутациям. В результате этого клетка может погибнуть или подвергнуться малигнизации.
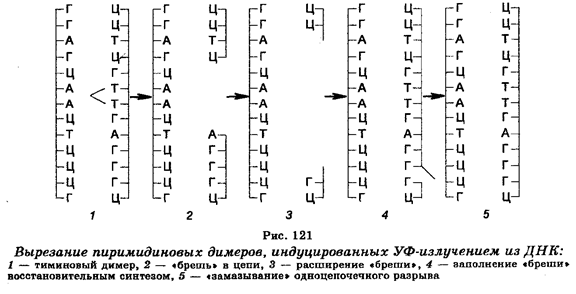
Один их механизмов восстановления повреждений ДНК действует у многих видов организмов, включая человека, и состоит в том, что экспонирование в видимом свете клеток, предварительно обработанных УФ-излучением, приводит к снижению летального эффекта в несколько раз, т. е. к фотореактивации функций облученных клеток. Это реактивирующее действие видимого света связано с расщеплением (мономеризацией) пиримидиновых димеров, причем этот процесс обеспечивается светозависимыми фотореактивирующими ферментами. Второй механизм удаления димеров пиримидиновых оснований из ДНК облученных клеток получил название темновой репарации или вырезания-восстановления. Так же, как и фотореактивация, он представляет собой ферментативный процесс, но более сложный, притом проходящий в темноте (рис. 121). Этот механизм заключается в том, что тиминовые димеры с помощью ДНК-репариру-ющих нуклеаз, осуществляющих гидролиз фосфодиэфирных скелетных связей между нуклеотидами с повреждениями (со стороны 5' от тиминового димера) и нормальной частью молекулы ДНК, подвергаются «вырезанию» из цепи ДНК, в которой после этого остаются бреши. Затем происходит «залатывание» брешей восстановительным синтезом ДНК при участии ДНК-полимеразы, связывающейся с 3'-концом поврежденной цепи ДНК, и использовании противоположной (нормальной) цепи в качестве шаблона. Удаление пиримидиновых димеров из ДНК облученных клеток путем «вырезания» и «залатывания» брешей завершается смыканием вновь реплицированного участка ДНК с соседними поврежденными участками и «замазыванием» («сшиванием») сахарофосфатных скелетных связей с помощью ДНК-лигазы. Таким образом, реализация этого механизма обеспечивается тремя репарирующими ферментами.
Третий механизм восстановления повреждений ДНК называют пострепликационным, или рекомбинационным восстановлением (рис. 122). Он заключается в том, что синтез ДНК в УФ-облученных клетках идет с нормальной скоростью вдоль хромосомы лишь до димера, перед которым он замедляется на несколько секунд, после чего начинается вновь, но уже на другой стороне димера. Поскольку ДНК-полимераза перескакивает через димер, то в дочерней цепи ДНК образуется брешь. Вследствие этого район, содержащий димер в одном дуплексе, будет интактным в сестринском дуплексе, т. е. в дочерних молекулах ДНК одна цепь будет содержать пиримидиновые димеры, тогда как другая будет иметь бреши, которые фактически являются вторичными повреждениями. Следовательно, район, содержащий димеры в одном дуплексе, полностью сохраняется в сестринском дуплексе. Этот процесс заканчивается рекомбинацией вдоль молекулы ДНК после ее репликации, при которой дочерняя цепь, несущая в каком-либо участке брешь, спаривается другой дочерней цепью (комплементарной), несущей брешь в другом месте. Это спаривание позволяет восстановительный синтез, который обеспечивает восстановление правильной последовательности района в каждой бреши. В качестве шаблона используется соответствующий интактный район другой дочерней цепи. Рекомбинационные события на уровне каждой бреши приводят к реконструкции интактной молекулы ДНК, способной к дальнейшей репликации. Рекомбинационное восстановление ДНК обеспечивается рядом белков-рекомбиназ.
 В ходе эволюции у клеток выработалась способность синтезировать репарирующие ферменты, которые синтезируются, когда начинают возникать повреждения ДНК. Например, у Е. coli открыта так называемая SOS-репарация, которая заключается в том, что любое повреждение ДНК, сопровождающееся нарушением ее репликации, ведет к усилению транскрипции большого количества генов (более 15), кодирующих репарирующие белки. Этот процесс сопровождается повышением выживаемости клеток. Известно также, что существуют репарирующие системы, которые активируются, если в ДНК образуются метилированные нуклеотиды. Подобные индуцированные системы репарации существуют, вероятно, и у эукариотических клеток.
В ходе эволюции у клеток выработалась способность синтезировать репарирующие ферменты, которые синтезируются, когда начинают возникать повреждения ДНК. Например, у Е. coli открыта так называемая SOS-репарация, которая заключается в том, что любое повреждение ДНК, сопровождающееся нарушением ее репликации, ведет к усилению транскрипции большого количества генов (более 15), кодирующих репарирующие белки. Этот процесс сопровождается повышением выживаемости клеток. Известно также, что существуют репарирующие системы, которые активируются, если в ДНК образуются метилированные нуклеотиды. Подобные индуцированные системы репарации существуют, вероятно, и у эукариотических клеток.
У человека известен синдром «ксеродерма пигментозум», который наследуется в качестве хромосомного рецессивного признака и который характеризуется чрезвычайной чувствительностью кожи к солнечному свету, в результате чего она подвергается избыточной пигментации, а затем часто происходит и малигнизация кожных клеток, т. е. развивается кожный рак. Возникновение этого синдрома связано с дефектом способности вырезать тиминовые димеры из ДНК. Известен также синдром Блума, наследуемый в качестве рецессивного признака и заключающийся в повышенной чувствительности индивидов к солнечному свету. Этот синдром связан также с увеличением геномной нестабильности в виде повышения сестринских хроматидных обменов, хромосомных аберраций в геномах больных, повышения риска развития у них всех типов рака и, самое главное, с дефектом восстановления их ДНК. Как для синдрома «ксеродерма пигментозум», так и для синдрома Блума характерна иммунологическая недостаточность у больных.
Нормальные повреждения ДНК, индуцированные солнечным светом (УФ-компонентом), восстанавливаются «вырезанием-восстановлением».
Некоторые из потенциально летальных или вторичных повреждений, индуцируемых рентгеновским излучением, могут быть восстанавливаемыми посредством рекомбинации или какого-либо другого механизма, в котором участвуют ферменты-рекомбиназы. Предполагается также, что в отличие от повреждений ДНК, индуцированных УФ-излучением, повреждения, индуцированные рентгеновским излучением, подвержены восстановлению (через рекомбинацию) еще до первой постлучевой репликации.
Повреждения, вызываемые в ДНК химическими мутагенами, также восстанавливаются с помощью того или иного механизма. Каждый из механизмов восстановления ДНК является, по существу, системой защиты ДНК. В то же время восстановление ДНК часто сопровождается ошибками, проявляющимися в форме мутаций.
Обобщая значение репарационных механизмов в жизни клеток, можно заключить, что репарация повреждений ДНК обеспечивает поддержание стабильности генов, причем она основана на наличии двух цепей в ДНК. Именно благодаря этому повреждения в одной цепи могут репарироваться за счет информации неповрежденной цепи. Однако ДНК, вопреки тому, что она является хранителем генетической информации, все же обладает ограниченной химической стабильностью. В клетках с довольно высокой частотой встречается гидролиз, окисление и неэнзиматическое метилирование ДНК. Эти реакции взаимодействуют с восстановлением ДНК. Предполагают, что спонтанный распад ДНК, вероятно, является главным фактором в спонтанном мутагенезе, карциногенезе и наступлении старения организмов. Таким образом, ДНК представляется противоречивой структурой. С одной стороны, она очень консервативна в плане ее стабильности, с другой стороны, она очень подвержена распаду.

