




В 90-х годах XX в. предложена рабочая классификация наследственных болезней человека, включающая:
1) синдромы, обусловленные хромосомными аномалиями (хромосомные болезни);
2) болезни, вызванные мутацией отдельного гена (генные или менделевские болезни);
3) мультифакториальные заболевания (МФЗ) как результат взаимодействия генетических и средовых факторов (болезни с наследственным предрасположением);
4) болезни с нетрадиционным типом наследования;
5) генетические болезни соматических клеток (новообразования, старение, аутоиммунные болезни).
Хромосомные болезни
Эта группа заболеваний обусловлена изменением структуры отдельных хромосом или их количества в кариотипе (см. разд. 4.2.1; 4.2.2; 6.4.3.6). Как правило, при таких мутациях наблюдается дисбаланс наследственного материала, который и ведет к нарушению развития организма. У человека описаны геномные мутации по типу полиплоидии, которые редко наблюдаются у живорожденных, а в основном обнаруживаются у абортированных эмбрионов и плодов и у мертворожденных. Основную часть хромосомных болезней составляют анэуплоидии, причем моносомии по аутосомам у живорожденных встречаются крайне редко. Большинство из них касаются 21-й и 22-й хромосом и чаще обнаруживаются у мозаиков, имеющих одновременно клетки с нормальным и мутантным кариотипом. Достаточно редко обнаруживается моносомия и по Х-хромосоме (синдром Шерешевского — Тернера).
В отличие от моносомии трисомии описаны по большому числу аутосом: 8, 9, 13, 14, 18, 21, 22-й и Х-хромосоме, которая может присутствовать в кариотипе в 4—5 экземплярах, что вполне совместимо с жизнью.
Структурные перестройки хромосом также, как правило, сопровождаются дисбалансом генетического материала (делеции, дупликации). Степень снижения жизнеспособности при хромосомных аберрациях зависит от количества недостающего или избыточного наследственного материала и от вида измененной хромосомы.
К настоящему времени описано около 100 клинико-цитогенетических синдромов, в основе которых лежат различные хромосомные аномалии.
Хромосомные изменения, приводящие к порокам развития, чаще всего привносятся в зиготу с гаметой одного из родителей при оплодотворении. При этом все клетки нового организма будут содержать аномальный хромосомный набор и для диагностики такого заболевания достаточно проанализировать кариотип клеток какой-нибудь ткани.
Если хромосомные нарушения возникают в одном из бластомеров во время первых делений зиготы, образующейся из нормальных гамет, то развивается мозаичный организм, большая или меньшая часть клеток которого несет нормальный хромосомный набор. Диагностика мозаичных форм хромосомных болезней отличается большей трудоемкостью и требует изучения кариотипа большого числа клеток из разных тканей.
Для определения вероятности появления хромосомной болезни в потомстве в семьях, уже имеющих больных детей, важно установить, является ли это хромосомное нарушение заново возникшим или оно унаследовано от предыдущего поколения. Чаще родители человека с хромосомным заболеванием имеют нормальный кариотип, а появление больного потомства является результатом мутации, возникшей в одной из гамет. В этом случае возможность повторного хромосомного нарушения у детей в данной семье маловероятна и не превосходит таковой в целом для популяции. Вместе с тем описано немало семей, в которых наблюдается предрасположение, например, к нерасхождению хромосом.
В случае наследуемых хромосомных болезней в соматических клетках родителей обнаруживаются хромосомные или геномные мутации, которые могут передаваться их зрелым половым клеткам в ходе гаметогенеза. Передают потомству хромосомные нарушения обычно фенотипически нормальные родители, являющиеся носителями сбалансированных хромосомных перестроек — реципрокных транслокаций, робертсоновских транслокаций или перицентрических инверсий. У носителей такого рода хромосомных перестроек с определенной вероятностью образуются нормальные гаметы, а также гаметы, несущие сбалансированную перестройку, и половые клетки с нарушенным балансом генов в геноме (рис. 6.22).
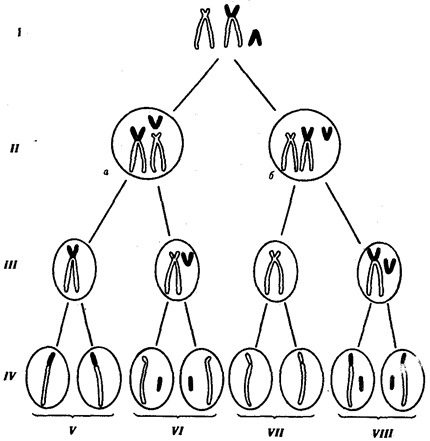
Рис. 6.22. Вероятность образования нормальных и аномальных гамет у носителей сбалансированной хромосомной перестройки
Показана робертсоновская транслокация 21-й хромосомы (окрашена) на одну из акродентрических хромосом (не окрашена);
I — кариотип носителя сбалансированной хромосомной перестройки, II — варианты (а, б) расположения бивалентов в экваториальной плоскости веретена деления (метафаза I), III — результат 1-го редукционного деления мейоза; IV — результат 2-го эквационного деления мейоза; V — гаметы со сбалансированной хромосомной перестройкой; VI — нормальные гаметы; VII — гаметы, не имеющие 21-й хромосомы; VIII — гаметы, содержащие две 21-е хромосомы (VII и VIII — гаметы с несбалансированным геномом)
Возможность наследования хромосомных аномалий делает необходимым анализ кариотипа родителей, уже имеющих больных детей, и пренатальную диагностику развивающегося внутриутробно плода для исключения вероятности повторного рождения ребенка с хромосомной болезнью.
Фенотипическое проявление различных хромосомных и геномных мутаций характеризуется ранним и множественным поражением различных систем органов. Типичными являются задержка общего физического и умственного развития, отклонения в строении скелета, в частности мозгового и лицевого черепа, пороки развития сердечно-сосудистой, мочеполовой, нервной систем, нарушения в биохимическом, гормональном и иммунологическом статусе организма. Хромосомные болезни, как правило, характеризуются сочетанием многих врожденных пороков. Для них также характерны многообразие и вариабельность фенотипических проявлений. Наиболее специфические проявления хромосомных заболеваний связаны с дисбалансом по относительно небольшому фрагменту хромосомы. Так, фенотипическое проявление синдрома Дауна наблюдается в случае трисомии всего лишь по небольшому сегменту длинного плеча 21-й хромосомы. Картина синдрома «кошачьего крика» развивается при утрате участка короткого плеча 5-й хромосомы. Дисбаланс по значительному объему хромосомного материала делает фенотипическую картину менее специфической.
Специфичность проявления хромосомного заболевания определяется изменением содержания определенных структурных генов, кодирующих синтез специфических белков. Так, при болезни Дауна обнаружено повышение в 1,5 раза активности фермента супероксид-дисмутазы I, ген которого располагается в 21-й хромосоме и представлен у больных в трехкратной дозе. Эффект «дозы гена» обнаружен более чем для 30 генов, локализованных в разных хромосомах человека.
Полуспецифические симптомы проявления хромосомных болезней связаны в значительной мере с дисбалансом генов, представленных многими копиями, которые контролируют ключевые процессы в жизнедеятельности клеток и кодируют, к примеру, структуру рРНК, тРНК, гистонов, рибосомальных белков, актина, тубулина.
Неспецифические проявления при хромосомных болезнях связывают с изменением содержания гетерохроматина в клетках, который оказывает влияние на нормальное течение клеточного деления и роста, формирование в онтогенезе количественных признаков, определяемых полигенами.
6.4.1.2. Генные ( или менделевские ) болезни
К указанным заболеваниям относятся моногенно обусловленные патологические состояния, наследуемые в соответствии с законами Менделя.
В зависимости от функциональной значимости первичных продуктов соответствующих генов генные болезни подразделяют на наследственные нарушения ферментных систем (энзимопатии), дефекты белков крови (гемоглобинопатии), дефекты структурных белков (коллагеновые болезни) и генные болезни с невыясненным первичным биохимическим дефектом.
Энзимопатии. В основе энзимопатии лежат либо изменения активности фермента, либо снижение интенсивности его синтеза. У гетерозигот-носителей мутантного гена присутствие нормального аллеля обеспечивает сохранение около 50% активности фермента по сравнению с нормальным состоянием. Поэтому наследственные дефекты ферментов клинически проявляются у гомозигот, а у гетерозигот недостаточная активность фермента выявляется специальными исследованиями.
В зависимости от характера нарушения обмена веществ в клетках среди энзимопатий различают следующие формы.
1. Наследственные дефекты обмена углеводов (галактоземия — нарушение метаболизма молочного сахара — лактозы; мукополисаха-ридозы — нарушение расщепления полисахаридов).
2. Наследственные дефекты обмена липидов и липопротеинов (сфинголипидозы — нарушение расщепления структурных липидов; нарушения обмена липидов плазмы крови, сопровождающиеся увеличением или снижением в крови холестерина, лецитина).
3. Наследственные дефекты обмена аминокислот (фенилкетонурия —нарушение обмена фенилаланина (см. разд. 4.1); тирозиноз— нарушение обмена тирозина; альбинизм — нарушение синтеза пигмента меланина из тирозина и др.).
4. Наследственные дефекты обмена витаминов (гомоцистинурия — развивается как результат генетического, дефекта кофермента витаминов В6 и B12, наследуется по аутосомно-рецессивному типу).
5. Наследственные дефекты обмена пуриновых и пиримидиновых азотистых оснований (синдром Леша — Найяна, связанный с недостаточностью фермента, который катализирует превращение свободных пуриновых оснований в нуклеотиды, наследуется по Х-сцепленному рецессивному типу).
6. Наследственные дефекты биосинтеза гормонов (адреногенитальный синдром, связанный с мутациями генов, которые контролируют синтез андрогенов; тестикулярная феминизация, при которой не образуются рецепторы андрогенов).
7. Наследственные дефекты ферментов эритроцитов (некоторые гемолитические несфероцитарные анемии, характеризующиеся нормальной структурой гемоглобина, но нарушением ферментной системы, участвующей в анаэробном (бескислородном) расщеплении глюкозы. Наследуются как по аутосомно-рецессивному, так и по Х-сцепленному рецессивному типу).
Гемоглобинопатии. Это группа наследственных заболеваний, вызываемых первичным дефектом пептидных цепей гемоглобина и связанным с этим нарушением его свойств и функций. К ним относят метгемоглобинемии, эритроцитозы, серповидно-клеточную анемию, талассемии (см. § 4.1).
Коллагеновые болезни. В основе возникновения этих заболеваний лежат генетические дефекты биосинтеза и распада коллагена — важнейшего структурного компонента соединительной ткани. К этой группе относят болезнь Эллерса — Данлоса, характеризующуюся большим генетическим полиморфизмом и наследующуюся как по аутосомно-доминантному, так и по аутосомно-рецессивному типу, болезнь Марфана, наследующуюся по аутосомно-доминантному типу, и ряд других заболеваний.

Рис. 6.23. Больные с ахондроплазией
Наследственные болезни с невыясненным первичным биохимическим дефектом. К этой группе принадлежит подавляющее большинство моногенных наследственных болезней. Наиболее распространенными являются следующие.
1. Муковисцидозы — встречаются с частотой 1:2500 новорожденных; наследуются по аутосомно-рецессивному типу. В основе патогенеза заболевания —наследственное поражение экзокринных желез и железистых клеток организма, выделение ими густого, измененного по составу секрета и связанные с этим последствия.
2. Ахондроплазия — заболевание, в 80—95% случаев обусловленное вновь возникшей мутацией; наследуется по аутосомно-доминантному типу; встречается с частотой приблизительно 1:100 000. Это заболевание костной системы, при котором наблюдаются аномалии развития хрящевой ткани преимущественно в эпифизах трубчатых костей и костях основания черепа (рис. 6.23).
3. Мышечные дистрофии (миопатии) — заболевания, связанные с поражением поперечно-полосатых и гладких мышц. Различные формы характеризуются разным типом наследования. Например, прогрессирующая псевдогипертрофическая миопатия Дюшена наследуется по Х-сцепленному рецессивному типу и проявляется преимущественно у мальчиков в начале первого десятилетия жизни. Известна мышечная псевдогипертрофическая дистрофия, наследующаяся по аутосомно-рецессивному типу, которая начинает развиваться во второй половине первого десятилетия жизни и встречается с одинаковой частотой у обоих полов. Мышечная дистрофия плечевого и тазового пояса: наследуется по аутосомно-доминантному типу и т.д.
Генетическое многообразие генных болезней. Изучение наследственных заболеваний у человека свидетельствует о том, что нередко сходное фенотипическое проявление болезни бывает обусловлено несколькими различными мутациями. Это явление впервые было описано в 30-х гг. С. Н. Давиденковым и названо генетической гетерогенностью наследственных заболеваний. Генетическая гетерогенность наследственных болезней может быть обусловлена мутациями разных генов, кодирующих ферменты одного метаболического пути, а также мутациями одного и того же гена, приводящими к появлению разных его аллелей.
Среди рассмотренных выше наследственных болезней особенно высокой степенью генетического полиморфизма отличаются мукопо-лисахаридозы, генетическая разнородность которых объясняется множественными мутациями в 11—12 генах, связанных общей функцией расщепления полисахаридов. Большой генетической гетерогенностью характеризуется врожденная аутосомно-рецессивная форма глухоты, при которой различают не менее 35 генетически различных вариантов с фенотипически сходным проявлением.
Большие перспективы в расшифровке наследственной гетерогенности генных болезней открываются в связи с применением молекулярно-генетических методов их прямого анализа с помощью ДНК-зондов.
Клиническое многообразие наследственных болезней. Разнообразие клиники наследственных болезней проявляется в различии времени начала заболевания, в спектре и степени выраженности симптомов, в течении и исходе у разных больных. Например, наследуемая по аутосомно-доминантному типу хорея Гентингтона, при которой поражаются базальные ганглии головного мозга, клинически начинает проявляться в виде непроизвольных движений в разном возрасте, но чаще в 40—45 лет. С временем начала клинического проявления связана и тяжесть течения заболевания (см. 6.4.1.4).
О клиническом полиморфизме можно говорить лишь в отношении генетически определенной наследственной формы. Причины клинического полиморфизма могут быть как генетическими, так и средовы-ми. К генетическим причинам можно отнести действие генов-модификаторов на проявление патологически измененного гена и сложную систему разнообразных взаимодействий между ним и другими генами. Кроме того, разнообразие клинического проявления наследственных заболеваний может зависеть от факторов среды, в которой развивается организм и которая влияет на проявление патологически измененных генов.

