




В рамках моногенного наследования выделяют:
• аутосомно-доминантный тип (на одной из двух аутосом расположен доминантный ген);
• аутосомно-рецессивный тип (на одной из двух аутосом расположен рецессивный ген);
• Х-сцепленный доминантный тип (на Х-хромосоме расположен доминантный ген);
• Х-сцепленный рецессивный тип (на Х-хромосоме расположен рецессивный ген);
• Y-сцепленный тип или голандрическое наследование (ген расположен на Y-хромосоме).
При однородительской дисомии обе гомологичные хромосомы происходят от одного родителя (т. е. хромосома другого родителя не наследуется). Возможный механизм дисомии — элиминация лишней хромосомы у плода с трисомией на ранних стадиях эмбриогенеза. Болезнь проявляется в том случае, если элиминируется лишняя хромосома, происходящая из нормальной гаметы.
1. У 20—30% больных с синдромом Прадера—Вилли, имеющих по данным цитогенетического исследования нормальный кариотип, с помощью молекулярно-биологических методов обнаруживается дисомия материнской 15-й хромосомы. Отцовская 15-я хромосома у таких больных отсутствует.
2. Однородительские дисомии были найдены при муковисцидозе и других моногенных заболеваниях, когда оба мутантных аллеля наследовались от одного родителя. В таких случаях дисомия имитирует аутосомно-рецессивное наследование.
3. Предполагают, что однородительская дисомия является причиной внутриутробной задержки развития, умственной отсталости и микроцефалии. Эти предположения подтверждены молекулярно-биологическими исследованиями.
В норме у человека имеется 46 хромосом, которые представлены 22 парами аутосом и одной парой половых хромосом — XX у женщин и XY — у мужчин. Из пары одинаковых (гомологичных) хромосом набора ребенок всегда наследует одну хромосому от матери и одну — от отца. Однако в результате однородительской дисомии — ОРД (англ. — Uniparental Disomy — UPD) потомок ошибочно наследует две гомологичные хромосомы от одного родителя — либо от матери (материнская ОРД — Орд мат, или Update), либо от отца (отцовская ОРД — ОРДотц, или UPDpat) и не имеет ни одной копии указанной гомологичной хромосомы от другого родителя. При этом в геноме потомка все остальные хромосомы набора имеют биродительское происхождение, а в целом кариотип сохраняет 46 хромосом и является, таким образом, псевдонормальным. ОРД могут возникать в результате нарушения процесса расхождения хромосом в мейозе в ходе образования мужских и женских половых клеток или в митотически делящихся клетках зиготы на ранних этапах развития зародыша по любой из 22 аутосом, а также и по половым хромосомам набора.
Существуют два типа ОРД:
а) изодисомия — наследование двух редупликационных копий одной из хромосом, возникающее при нерасхождении хромосом во II делении мейоза. В результате такого события обе хромосомы имеют идентичную последовательность ДНК с полной гомозиготизацией локализованных в них генов;
б) гетеродисомия — наследование потомком двух разных гомологов от одного родителя в результате нерасхождения хромосом в I мейотическом делении. В данном случае потомок наследует неидентичные по последовательности ДНК гомологичные хромосомы.
98.Полигенные или мультифакториальные заболевания(псориаз,сахарный диабет,шизофрения)
Плигенные болезни (ранее - заболевания с наследственной предрасположенностью) обусловлены как наследственными факторами, так и, в значительной степени, факторами внешней среды. Кроме того, они связаны с действием многих генов, поэтому их называют также мультифакториальными. К наиболее часто встречающимся мультифакториальным болезням относятся: ревматоидный артрит, ишемическая болезнь сердца, гипертоническая и язвенная болезни, цирроз печени, сахарный диабет, бронхиальная астма, псориаз, шизофрения и др. Полигенные заболевания тесно связаны с врождёнными дефектами метаболизма, часть из которых может проявляться в виде метаболических заболеваний
Распространение полигенных наследственных заболеваний
Эта группа болезней в настоящее время составляет 92% от общего числа наследственных патологий человека. С возрастом частота заболеваний возрастает. В детском возрасте процент больных составляет не менее 10 %, а в пожилом - 25-30 %.
Распространение мультифакториальных болезней в разных популяциях человека может значительно варьировать, что связано с различием генетических и средовых факторов. В результате генетических процессов, происходящих в человеческих популяциях (отбор, мутации, миграции, дрейф генов), частота генов, определяющих наследственную предрасположенность, может возрастать или уменьшаться вплоть до полной их элиминации.
Особенности полигенных болезней Клиническая картина и тяжесть течения мультифакториальных болезней человека в зависимости от пола и возраста очень различны. Вместе с тем, при всем их разнообразии, выделяют следующие общие особенности:
Высокая частота заболеваний в популяции. Так, шизофренией болеют около 1% населения, сахарным диабетом — 5%, аллергическими заболеваниями — более 10%, гипертонией — около 30%.
Клинический полиморфизм заболеваний варьирует от скрытых субклинических форм до ярко выраженных проявлений.
Особенности наследования заболеваний не соответствуют менделевским закономерностям. Степень проявления болезни зависит от пола и возраста больного, интенсивности работы его эндокринной системы, неблагоприятных факторов внешней и внутренней среды, например, нерационального питания и др.
Генетическое прогнозирование полигенных болезней Генетический прогноз при мультифакториальных заболеваниях зависит от следующих факторов: чем ниже частота болезни в популяции, тем выше риск для родственников пробанда; чем сильнее степень выраженности болезни у пробанда, тем больше риск развития болезни у его родственников; риск для родственников пробанда зависит от степени родства с пораженным членом семьи; риск для родственников будет выше, если пробанд относится к менее поражаемому полу.
Полигенная природа болезней с наследственной предрасположенностью подтверждается с помощью генеалогического, близнецового и популяционно-статистического методов. Достаточно объективен и чувствителен близнецовый метод. С помощью близнецового метода показана наследственная предрасположенность к некоторым инфекционным заболеваниям (туберкулез, полиомиелит) и многим распространенным болезням (ишемическая болезнь сердца, ревматоидный артрит, сахарный диабет, язвенная болезнь, шизофрения и др.).
Гипергликемия – увеличение содержания глюкозы в крови. Может носить физиологический характер в случае приема богатой углеводами пищи (алиментарная гипогликемия) или в результате одномоментной физической нагрузки: адреналин, глюкокортикостероиды и катехоламины усиливают глюконеогенез и распад гликогена. Физиологические гипергликемии носят кратковременный характер. Патологические типы гипергликемий обусловлены эндокринными расстройствами, в частности нарушением оптимального соотношения между секрецией гормонов гипер- и гипогликемического действия. Наиболее распространенная форма патологической гипергликемии – сахарный диабет, обусловленный дефицитом инсулина. В норме продукцию инсулина секреторными клетками поджелудочной железы стимулирует глюкоза, лейцин и глутаминовая кислота, кетоновые тела и некоторые жирные кислоты. Дефицит инсулина может быть обусловлен генетическими нарушениями синтеза этого гормона или заболеваниями поджелудочной железы (панкреатит, панкреонекроз) – вторичный диабет. При дефиците инсулина развивается гипергликемия, которая вызвана нарушением транспорта глюкозы в клетки. Кроме того, дефицит инсулина приводит к стимуляции глюконеогенеза и гликогенолиза. Глюкозурия (глюкоза в моче) связана с нарушением инсулинзависимой реабсорбции глюкозы. Кетонемия и кетонурия обусловлены тем, что дефицит глюкозы в клетках активирует окисление жирных кислот где образуется большое количество ацетил-КоА. Он не может быть полностью использован в ЦТК и часть его идет на синтез кетоновых тел. Накопление их в крови приводит к кетоацидозу, т.е. к смещению кислотно-основного состояния организма в кислую сторону. Помимо сахарного диабета гипергликемии могут быть обусловлены повышенной секрецией соматотропного гормона и АГКТ, катехоламинов и глюкокортикоидов как результат заболеваний гипоталамуса и надпочечников. Гипогликемия может носить физиологический характер вслед за алиментарной гипергликемией как результат компенсаторного выброса инсулина.
Патологическая гипогликемия может быть результатом: гиперинсулинемии; недостаточностью ферментов расщепляющих дисахариды в кишечнике; заболеваний печени с торможением гликогенобразования и глюконеогенеза; дефицита глюкокортикоидов; гипоксии. Стоит остановиться на особенностях обмена глюкозы в клетках при гипоксии: 1) при дефиците кислорода в клетках метаболизм становится анаэробным, что приводит к накоплению молочной кислоты; 2) для обеспечения клетки энергией активируется гликолиз, что приводит к накоплению лактата, в меньшей степени пирувата и дефициту в крови глюкозы; 3) дефицит кислорода, как конечного акцептора ЭТЦ и пирувата, как основного субстрата ЦТК замедляет активность работы этих участков метаболизма глюкозы, что приводит к резкому снижению в клетках концентрации АТФ; 4) далее развиваются патологические процессы общего характера (прекращение работы K+-Na+-насоса, активация процессов ПОЛ) и пр. Таким образом, этиология гипер- и гипогликемий может носить физиологический так и патологический характер, а патогенез обязательно включает как нейроэндокринные, так и молекулярные нарушения, где последние чаще всего носят наследственный характер.
Псориаз, чешуйчатый лишай — хроническое неинфекционное заболевание, дерматоз, поражающий в основномкожу[1][2][3]. В настоящее время предполагается аутоиммунная природа этого заболевания[4]. Обычно псориаз проявляется образованием красных, чрезмерно сухих, приподнятых над поверхностью кожи пятен — так называемых папул, которые сливаются между собой, образуя бляшки. Эти папулы являются по своей природе участками хронического воспаления и избыточной пролиферации лимфоцитов, макрофагов и кератиноцитов кожи, а также избыточного ангиогенеза (образования новых мелких капилляров) в подлежащем слое кожи.
От псориаза страдает около 4% населения земного шара. Он может развиваться в любом возрасте с момента рождения и до глубокой старости, однако больше всего псориаз «любит» молодых. Об этом свидетельствует тот факт, что 70% пациентов, заболевают псориазом в возрасте до 20 лет.
Псориаз — это ненормальная реакция организма на внешние раздражители, при которой на отдельных участках тела верхний слой кожи отмирает гораздо быстрее, чем в норме. Если обычно цикл деления и созревания клеток кожи происходит за 3-4 недели, то при псориазе этот процесс протекает всего за 4-5 дней.
В настоящее время псориаз считают наследственным мультифакториальным заболеванием: в основе болезни лежит не одна, а целый комплекс причин – иммунологические сдвиги, нарушение обмена веществ, сопутствующие эндокринные и неврологические расстройства. При этом можно сказать точно: псориаз — не инфекционное, а значит и не заразное заболевание.
Причины возникновения псориаза до сих пор окончательно не найдены. На этот счет существуют различные теории.
По одной из теорий, существует два типа псориаза:
· Псориаз I типа вызывается передающимися по наследству поломками иммунной системы. Этой формой псориаза болеют около 65% людей, причем заболевание проявляется в молодом возрасте, от 18 до 25 лет.
· Псориаз II типа проявляется у людей старше 40 лет. При этом типе псориаз не передается по наследству и не связан с поломками в клетках иммунной системой. Причем в отличие от псориаза I типа, который предпочитает кожу, псориаз II типа чаще поражает ногти и суставы.
Согласно другой теории, причиной псориаза являются исключительно нарушения иммунитета, вызванные различными факторами: это может быть стресс, или инфекционные заболевания, или холодный климат, или неправильное питание. Например, отмечено, что алкоголь может спровоцировать обострение псориаза — особенно это относится к пиву, шампанскому, крепким спиртным напиткам.
Шизофрения — это психическое заболевание, которое имеет длительное течение и сопровождается рассогласованностью психических процессов, моторики и нарастающими изменениями личности.
Чаще всего болезнь начинается в возрасте от 15 до 25 лет и имеет прогрессирующее течение.
Причины заболевания
Ведущую роль в возникновении шизофрении имеет наследственная предрасположенность. Внешние факторы (психические травмы, перенесенные заболевания, травмы головы и пр.) имеют вторичное значение, выступая только в роли активатора психопатологического процесса.
Симптомы шизофрении
- слуховые псевдогаллюцинации (например, кто-то вкладывает больному свои мысли или отнимает у больного его мысли);
- бред воздействия (кто-то или что-то действует необычным способом, руководя мыслями, чувствами, поведением человека или подвергая опасности его здоровье);
- вербальные галлюцинации (больной слышит голоса);
- стойкий, вычурный бред (контакт с инопланетянами, потусторонним миром и т.д.)
- уплощение, неадекватность и обеднение эмоций.
Могут также встречаться симптомы кататонии (ступор, возбуждение), эпизоды неуправляемого потока мыслей, разорванность речи, признаки апатико-абулического синдрома сочетание безволия с безразличием и утратой желаний.
Диагностика шизофрении
Диагностикой и лечением шизофрении занимается врач-психиатр. В некоторых случаях для обследования пациента требуется госпитализация в психиатрическую больницу. Диагноз «шизофрения» ставится, как правило, при длительности заболевания не менее 6 месяцев.
99.Классификация наследственных болезней (по Ярыгину:генные хромосомные,мультифактор.,болезни с нетрадиц. типом насл-я,ген.болезни соматических клеток)
100.Генные болезни (фенилкетонурия, синдром Марфана, ахондроплазия,серповидно клеточная анемия)
Генные болезни- это разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями на генном уровне.
Классифиция генных болезней:
•по типу наследования генные болезни делятся на аутосомно-доминантные, аутосомно-рецессивные, Х-сцепленные доминантные и т.д.
•в зависимости от системы или органа, наиболее вовлеченного в патологический процесс генные болезни делятся на нервные, нервно-мышечные, кожные, глазные, опорно-двигательного аппарата, эндокринные, крови, легких, сердечно-сосудистой системы, мочеполовой системы, желудочно-кишечного тракта и др.
•по характеру метаболического дефекта генные болезни делятся на болезни, связанные с нарушением аминокислотного, углеводного, липидного, минерального обменов, обмена нуклеиновых кислот и др.
•самостоятельную группу составляют наследственно обусловленные заболевания, возникающие при несовместимости матери и плода по антигенам групп крови
Фенилкетонури́я (фенилпировиноградная олигофрения) — редкое наследственное заболевание группыферментопатий, связанное с нарушением метаболизма аминокислот, главным образом фенилаланина. Сопровождается накоплением фенилаланина и его токсических продуктов, что приводит к тяжёлому поражению ЦНС, проявляющемуся, в частности, в виде нарушения умственного развития.
Диагностика заболевания осуществляется биохимическими методами: ещё до развития клинической картины в моче определяется ФПВК, в крови - высокое содержание фенилаланина. В родильных домах обязательно проводится скрининг-тест на фенилкетонурию.
Синдром (болезнь) Марфана — - аутосомно-доминантная болезнь, причиной которой является мутация гена белка фибриллина (15q21). Описано более 10 вариантов мутаций в одном гене, что обусловливает крайнюю вариабельность синдрома. Семейные случаи составляют 75% всех наблюдений. Популяционная частота-1:10 000.. В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлиненные конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставов), наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.
Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30—40 годами[1], и смерть наступает вследствие расслаивающейся аневризмы аорты или застойной сердечной недостаточности. В странах с развитым здравоохранением больные успешно лечатся и доживают до преклонного возраста.
При СМ нарушен синтез белка фибриллина и соединительная ткань обладает повышенной растяжимостью. Симптоматика СМ многосистемная и разнообразная. Наиболее специфическими для диагностики СМ являются поражения опорно-двигательного аппарата, вывих хрусталика, пороки развития сердечно-сосудистой системы, эктазия твердой мозговой оболочки. Основные нарушения скелетно-мышечной системы: арахнодак-тилия, высокий рост, длинные конечности, деформация позвоночника (сколиоз, грудной лордоз, гиперкифоз), деформация передней стенки грудной клетки (вдавленная грудь, "куриная" грудь), ненормальная подвижность суставов, плоская стопа, высокое арковидное нёбо, недоразвитие вертлужной впадины, мышечная гипотония. Основные симптомы поражения глазной системы: вывих хрусталика, миопия, отслоение сетчатки, уплощение роговицы, увеличенная длина оси глазного яблока. В сердечно-сосудистой системе обнаруживаются: аортальная регур-гитация, аневризма восходящей части аорты, расслоение аорты, пролапс митрального клапана, кальцификация митрального отверстия, дизритмия. Для заболевания характерны паховые грыжи, спонтанный пневмоторакс, аномалии развития нервной системы.
Диагностические критерии СМ должны строго соблюдаться, так как ряд других врожденных дисплазий соединительной ткани могут быть приняты за СМ. Подтверждается диагноз наличием этого заболевания у родственников 1-й степени родства.
Серповидноклеточная анемия — это наследственная гемоглобинопатия, связанная с таким нарушением строениябелка гемоглобина, при котором он приобретает особое кристаллическое строение — так называемый гемоглобин S.Эритроциты, несущие гемоглобин S вместо нормального гемоглобина А, под микроскопом имеют характерную серпообразную форму (форму серпа), за что эта форма гемоглобинопатии и получила название серповидноклеточной анемии.
Серповидноклеточная анемия весьма распространена в регионах мира, эндемичных по малярии, причём больные серповидноклеточной анемией обладают повышенной (хотя и не абсолютной) врождённой устойчивостью к заражению различными штаммами малярийного плазмодия. Серповидные эритроциты этих больных также не поддаются заражению малярийным плазмодием в пробирке. Повышенной устойчивостью к малярии обладают и гетерозиготы-носители, которые анемией не болеют (преимущество гетерозигот), что объясняет высокую частоту этого вредного аллеля в африканских популяциях.
· Усталость и анемия
· Приступы боли
· Отек и воспаление пальцев рук и/или ног и артрит
· Бактериальные инфекции
· Тромбоз крови в селезенке и печени
· Лёгочные и сердечные травмы
· Язвы на ногах
· Асептический некроз
· Повреждение глаз
Симптомы серповидноклеточной анемии делятся на две основные категории. Из-за хрупкости красных клеток крови всегда наблюдается анемия, которая может привести к потере сознания, делает больного физически менее выносливым и может вызвать желтуху (связанную с чрезмерным распадом гемоглобина).
Почти невозможно описать «типичного пациента», страдающего серповидноклеточной анемией, поскольку симптомы и их тяжесть широко варьируют. Некоторые характерные особенности являются общими почти для всех пациентов с серповидноклеточной анемией
В периоды гемолитических кризисов отмечается резкое падение уровня гемоглобина, которое сопровождается высокой температурой и черным цветом мочи. У больных серповидной анемией меняется и внешний вид: отмечается высокий рост, худоба, удлиненность туловища, искривление позвоночника, башенный череп и измененные зубы.
Ахондроплазия — системное поражение скелета — врождённая болезнь, характеризующаяся нарушением энхондрального остеогенеза; проявляется карликовостью, короткими конечностями при обычной длине туловища, деформацией нижних конечностей и позвоночника и относительной макроцефалией.
Нарушение анатомических пропорций заметно уже при рождении: ребенок с ахондроплазией имеет относительно большую голову, короткие ручки и ножки. Лоб выпуклый, мозговая часть черепа увеличена, затылочные и теменные бугры выпирают. В отдельных случаях возможна гидроцефалия. Отмечаются нарушения строения лицевого скелета, возникшие вследствие неправильного развития костей основания черепа. Глаза пациентов с ахондроплазией широко расставлены, находятся глубоко в орбитах, около внутренних углов глаз есть дополнительные складочки. Нос седловидный, сплющенный, с широкой верхней частью, лобные кости заметно выступают вперед, верхняя челюсть также значительно выступает вперед над нижней. Язык грубый, небо высокое.
В настоящее время удовлетворительное лечение не известно, хотя и найдена причина заболевания — мутация в рецепторе ростового гормона. В связи с этой этиологией, человеческий гормон роста мало помогает больным ахондроплазией, при том, что помогает при некоторых других заболеваниях, которые без лечения приводят к аномально малому росту.
Другой метод увеличения роста при ахондроплазии — хирургическое удлинение костей, которое позволяет увеличить рост больного приблизительно на 24-28 см.
101.Хромосомные болезни(синдром Дауна,синдром Эдвардса,синдром Патау, синдром кошачьего крика)
Хромосомные болезни — наследственные заболевания, обусловленные изменением числа или структуры хромосом. К хромосомным относятся болезни, обусловленные геномными мутациями или структурными изменениями отдельных хромосом. Хромосомные болезни возникают в результате мутаций в половых клетках одного из родителей. Из поколения в поколение передаются не более 3—5 % из них. Хромосомными нарушениями обусловлены примерно 50 % спонтанных абортов и 7 % всех мёртворождений.
Все хромосомные болезни принято делить на две группы: аномалии числа хромосом и нарушения структуры хромосом.
Синдром Дауна (трисомия хромосомы 21). Впервые описан в 1866 году английским врачом Дауном. Наиболее часто встречающийся хромосомный синдром - популяционная частота составляет 1 случай на 600-700 новорожденных детей. Частота рождения детей с данным синдромом зависит от возраста матери и резко увеличивается после 35 лет. Цитогенетические варианты очень разнообразны, но около 95% случаев представлены простой трисомиеи 21 хромосомы, врезультате нерасхождения хромосом в мейозе у родителей. Наличие полиморфных молекулярно-генетических маркеров позволяет определить конкретного родителя и стадию мейоза в которой произошло нерасхождение (М1 - нерасхождениетомологичных хромосом 21 и М2 - нерасхождение хроматид). Несмотря на интенсивное изучение синдрома причинынерасхождения хромосом до настоящего времени не ясны. Этиологически важными факторами считаются внутри и внефолликулярное перезревание яйцеклетки, снижение числа или отсутствие хиазм в 1-м делении мейоза. Отмечены мозаичные формы синдрома (2%), робертсоновские транслокационные варианты (4%). Около 50% транслокационных форм наследуются от родителей и 50% являются мутациями de novo. Критическим сегментом, ответственным заформирование основных признаков синдрома, является область 21 q22.
Основными диагностическими признаками синдрома являются: типичное плоское лицо, монголоидный разрез глаз, эпикант, открытый рот, макроглоссия и аномалии зубов, короткий нос и плоская переносица, избыток кожи на шее, короткие конечности, поперечная четырех-пальцевая ладонная складка (обезьянья борозда). Из пороков внутренних органов часто отмечаются врожденные пороки сердца и желудочно-кишечного тракта, которые и определяют продолжительность жизни больных. Умственная отсталость обычно средней степени тяжести. Дети с синдромом Дауна часто ласковые и привязчивые, послушные и внимательные.
Клинико-цитогенетическая характеристика синдромов, связанных с аномалиями половых хромосом
Синдром Эдвардса (трисомия по хромосоме 18). Описан в 1960 году. Популяционная частота составляет 1 на 6500. Цитогенетически в большинстве случаев представлен целой трисомиеи 18 (гаметическая мутация одного из родителей, чаще по материнской линии). Кроме того, встречаются и мозаичные формы, а транслокации наблюдаются очень редко. Критическим сегментом, ответственным за формирование основных признаков синдрома, является сегмент 18q11. Клинических различий между цитогенетическими формами не обнаружено.
Дети с синдромом Эдвардса имеют малую массу тела при рождении. Основными диагностическими признакамисиндрома являются: долихоцефалия, гипертелоризм, низко посаженные аномальной формы уши, микрогнатия, микростомия, скошенный подбородок. Имеются аномалии развития конечностей: верхних - сгибательные деформациипальцев, перекрывание пальцев, сжатые пальцы рук, гипоплазия ногтей (особенно V пальца); нижних - короткий и широкий палец стопы, типичная форма стопы в виде качалки, кожная синдактилия стоп. Из внутренних пороков следует отметить комбинированные пороки сердечно-сосудистой системы, незавершенный поворот кишечника пороки развития почекчаще гидронефроз и подковообразная почка), крипторхизм. Отмечается задержка психомоторного развития, идиотия и имбецильность. Дети погибают, в основном, в возрасте до 1 года от осложнений, вызванных врожденными порока миразвития.
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 месяцев, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Синдром Патау - это хромосомная аномалия, синдром при котором у пациента есть дополнительная 13 хромосома, в связи с нерасхождением хромосом во время мейоза (также известный как трисомия 13 и трисомия D). Некоторые из них вызваны робертсоновской транслокацией. Дополнительная 13 хромосома нарушает нормальный ход развития ребенка, а именно возникновение дефектов сердца и почек, кроме других особенностей, характерных для синдрома Патау. Как и все болезни, которые вызваны нерасхождением (такие как синдром Дауна исиндром Эдвардса), риск возникновения этого синдрома у потомства увеличивается с возрастом матери при беременности (в среднем с 31 года). Синдром Патау поражает примерно 1 человека из 10000 новорожденных.
Для синдрома Патау характерны следующие диагностические признаки: микроцефалия, расщелина верхней губы и неба, низко посаженные деформированные ушные раковины, микрогения, полидактилия, флексорное положение пальцев рук, выпуклые ногти, поперечная ладонная складка, стопа-качалка. Из пороков внутренних органов отмечены врожденные пороки сердца (дефекты перегородок и крупных сосудов), незавершенный поворот кишечника, дивертикул Меккеля, поликистоз почек, удвоение мочеточника. Наблюдается крипторхизм, гипоплазия наружных половых органов, удвоениематки и влагалища. Глубокая идиотия. Дети, в основном, умирают в возрасте до 1 года, чаще в первые 2-3 месяца жизни.
Медицинское лечение детей с трисомией 13 планируется в каждом конкретном случае отдельно и зависит от индивидуального состояния пациента. Лечение синдрома Патау сосредоточивается на конкретных физических проблемах, с которыми рождается ребенок. Многие дети борются за выживание в первые несколько дней или недель своей жизни через тяжелые неврологические проблемы или сложные дефекты сердца. Хирургия может быть необходимой для устранения пороков сердца, заячьей губы и волчьей пасти. Для реализации своего потенциального развития пациентам необходима помощь логопеда, а также физическая и профессиональная терапия.
Синдром кошачьего крика (также известен под названиями: синдром делеции короткого плеча 5 хромосомы, 5р синдром или синдром Лежена) - редкое генетическое заболевание, связанное с отсутствием части 5 хромосомы. У больных детей с этим заболеванием (преимущественно, но нельзя сказать, что у всех) проявляется плач, который похож на кошачий крик, именно поэтому этот синдром получил название Cri-Du-Chat Syndrome, что происходит от французских слов (плач кошки или крик кота). Впервые болезнь описал Жером Лежен в 1963 году. Частота возникновения синдрома - 1 ребенок на 50000 родившихся, встречается во всех этнических групп и чаще ею болеют женщины, соотношение мужской и женской стати составляет 4:3.
Признаки и симптомы
Как уже было отмечено, синдром получил свое название из-за характерного плача детей (он аналогичен мявканнюкотенка, крика кошки), страдающих этим заболеванием. Это крик происходит из-за проблем с гортанью и нервной системой. Около 1/3 детей теряют эту особую характерную черту до 2 лет. Другими симптомами, которые указывают на заболевания синдромом кошачьего крика являются:
• проблемы с питанием в связи с трудностями при глотании и сосании;
• низкий вес ребенка при рождении и низкие темпы развития (в первую очередь физического);
• существенная задержка развития когнитивных, речевых функций и функций движения;
• проблемы с поведением, такие как: гиперактивность, агрессия, истерики и однообразные движения, постоянно повторяются;
• нетипичные черты лица, которые могут со временем исчезнуть или усилиться;
• чрезмерное, неконтролируемое слюноотделение;
• запоры.
Диагноз ставят на основании характерного детского крика и других характерных для этого заболевания симптомов.Генетическая консультация и генетическое тестирование могут быть предложены для семей в которых есть люди больные этим синдромом. Сердечные пороки часто требуют хирургической коррекции, поэтому часто необходима консультация детского кардиохирурга и специальная диагностика (эхо-кардиография).
102.Синдромы половых хромосом,механизмы их возникновения(синдром Шершевского-Тернера, Клайнфельтера и др)
Хромосомные болезни — наследственные заболевания, обусловленные изменением числа или структуры хромосом. К хромосомным относятся болезни, обусловленные геномными мутациями или структурными изменениями отдельных хромосом. Хромосомные болезни возникают в результате мутаций в половых клетках одного из родителей. Из поколения в поколение передаются не более 3—5 % из них. Хромосомными нарушениями обусловлены примерно 50 % спонтанных абортов и 7 % всех мёртворождений.
Все хромосомные болезни принято делить на две группы: аномалии числа хромосом и нарушения структуры хромосом.
1. Синдром Тернера — это клиническое проявление аномалии одной из X-хромосом у женщин. Синдром Тернера в 60% случаев обусловлен моносомией X-хромосомы (кариотип 45,X), в 20% случаев — мозаицизмом (например, 45,X/46,XX) и в 20% случаев — аберрацией одной из X-хромосом (например, 46,X[delXp–]). Распространенность синдрома Тернера, обусловленного полной моносомией X-хромосомы (45,X), среди детей, родившихся живыми, составляет 1:5000(у девочек 1:2500). Плоды с кариотипом 45,X спонтанно абортируются в 98% случаев. Синдром характеризуется множественными пороками развития скелета и внутренних органов. Важнейшие фенотипические признаки: низкорослость и дисгенезия или полное отсутствие гонад (на месте яичников находят недифференцированные соединительнотканные тяжи, не содержащие половых клеток и фолликулов). Другие признаки: короткая шея с крыловидными кожными складками, низкая линия роста волос на затылке, бочкообразная грудная клетка, нарушение пропорций лица, О-образное искривление рук (деформация локтевых суставов), X-образное искривление ног.
Симптомы синдрома Тернера - невысокий рост (130-150 см), бесплодие, лицо «сфинкса» из-за уменьшенного подбородка, деформированные ушные раковины. Другие часто встречающиеся врожденные отклонения - короткая шея, крыловидные складки на шее, недоразвитие скелета, пороки сердца и аномалии почек. Отсутствие или недоразвитие вторичных половых признаков.
Синдром Клайнфельтера (синдром XXY)
Это сочетание называется синдромом Клайнфельтера, или синдромом XXY - хромосомная болезнь, дисомия по X-хромосоме у мужчин. Сначала больные мальчики по внешнему виду ничем не отличаются от здоровых. Со временем у них отмечаются некоторые отклонения: небольшие размеры яичек, бесплодие, непропорционально длинные ноги. Кроме того, у многих наблюдается слабо выраженная умственная отсталость. В связи с тем, что при данном синдроме Х-хромосом больше, у больных происходит увеличение грудной железы.
Синдром XXX
Клинические проявления синдрома XXX весьма полиморфны, а у части пациентов с трисомией-Х вообще не обнаруживается каких-либо отклонений в физическом и психическом развитии. Вместе с тем одним из частых проявлений трисомии-Х является неглубокая умственная отсталость, которая отмечается у 75% больных. У многих больных с трисомией-Х наблюдаются задержка физического развития, негрубые диспластические признаки: эпикант, высокое твердое небо. Реже встречаются больные высокого роста. У некоторых пациентов отмечается бесплодие, обусловленное недоразвитием фолликулов.
Синдром XYY
В этом случае в клетках находится не одна, а две Y хромосомы. Рост у таких мужчин обычно выше среднего. У них отмечается слабо выраженная умственная отсталость, авторитарный тип характера, что можно объяснить избытком мужского гормона тестостерона в организме.
103.Электронные базы данных наследственных болезней (каталог МакКьюсика, Оксфордская мед база данных, СИНГЕН,ХРОДИС,МЕДГЕН-2000)
В настоящее время разработаны диагностические программы для использования врачами не генетических специальностей для постановки диагноза наледственно-больному человеку.
1) Каталог менделирующих признаков человека В. Мак-Кьюсика (OMIM)
Содержит около 11 тыс. наследственных болезней. OMIM предназначен для использования врачами и другими специалистами, имеющими непосредственное отношение к генетическим нарушениям, учеными-генетиками и студентами-медиками, биологами. OMIM также открыта для общего пользования, поэтому любые пользователи, ищущие информацию в базе данных, имеют возможность обращаться к ней для постановки диагноза и получения ответов на персональные вопросы. Ниже приведен логотип этой базы данных.
2) МЕДГЕН. каталог медецинской генетики с описанием около 2-3 тыс болезней
3)ХРОДИС (хромосомные дизморфии) каталог включающий все хромосомные болезни
4) OMD (Oxford Medical Database - Оксфордская медицинская база данных), которая имеет два подраздела - Лондонскую базу данных (London Dysmorphology Database — LDDB), включающую сведения более чем о 2300 нехромосомных синдромах и Лондонскую нейрогенетическую базу данных (London Neurogenetics Database — LNDB), содержащую сведения более чем о 2200 наследственных заболеваниях центральной и периферической нервной системы. Базы данных основаны на сведениях более чем из 1000 научных журналов и постоянно пополняются.
5) СИНГЕН - синдромы генетические - информационно диагностическая система, включающая 2000 синдромов и врожденных пороков развития человека.
104.Методы лабораторной диагностики наследственных болезней (цитогенетические, биохимические)
Лаборато́рная диагно́стика — совокупность методов, направленных на анализ исследуемого материала с помощью различного специализированного оборудования.
Существует 5 основных методов диагностики:
1) микроскопический — позволяет обнаружить возбудителя непосредственно в материале, взятом от больного. Для этого мазок окрашивают различными способами. Этот метод играет решающую роль при диагностике многих инфекционных заболеваний: туберкулеза, малярии, гонореи и др.;
2) бактериологический — заключается в посеве исследуемого материала на питательные среды. Этот метод позволяет выделить возбудителя в чистом виде и изучить его морфологические признаки, ферментативную активность и идентифицировать его;
3) биологический метод — осуществляют путем выделения возбудителя при заражении лабораторных животных, которые восприимчивы к данному заболеванию. Этот метод дорогостоящий, поэтому применяется ограниченно;
4) серологические методы исследования — основаны на выявлении специфических иммунных антител в сыворотке крови больного. Для этого используют различные иммунологические реакции: реакция Видаля (используется для выявления брюшного тифа);
5) аллергический метод — ставятся кожно-аллергические пробы, введение аллергена накожно или внутрикожно; используются для диагностики туберкулеза, туляремии, лепры и т. д.
Цитогенетический метод. С помощью данного метода можно изучать наследственный материал клетки: совокупность хромосом в целом (кариотипирование) или наличие и количество Х-хромосом (определение полового хроматина — число глыбок полового хроматина или телец Барра). Исследование проводится с помощью светового микроскопа (изготовление и изучение микропрепаратов).
Биохимические методы направлены на выявление биохимического фенотипа организма. Эти методы позволяют диагностировать наследственные болезни, обусловленные генными мутациями. Биохимические показатели (первичный белковый продукт гена, накопление патологических метаболитов внутри клетки) отражают сущность болезни более адекватно, чем клинические симптомы. С помощью биохимических методов описано более 1000 врожденных болезней обмена веществ. Наиболее распространенными среди таких заболеваний являются болезни, связанные с дефектами ферментов, структурных и транспортных белков. Дефекты ферментов устанавливают путем определения содержания в биологических средах (например, моче и крови) продуктов метаболизма, являющихся продуктом функционирования данного белка.
105. Методы медецинской генетики (генеалогический, близнецовый,дерматоглифика)
Медицинская генетика (или генетика человека, клиническая генетика, генопатология) — область медицины, наука, которая изучает явления наследственности и изменчивости в различных популяциях людей, особенности проявления и развития нормальных и патологических признаков, зависимость заболеваний от генетической предрасположенности и условий окружающей среды. Задачей медицинской генетики является выявление, изучение, профилактика и лечение наследственных болезней, разработка путей предотвращения воздействия негативных факторов среды на наследственность человека.
Генеалогический метод
Использование этого метода возможно в том случае, когда известны прямые родственники — предки обладателя наследственного признака (пробанда) по материнской и отцовской линиям в ряду поколений или потомки пробанда также в нескольких поколениях. При составлении родословных в генетике используется определенная система обозначений. После составления родословной проводится ее анализ с целью установления характера наследования изучаемого признака.
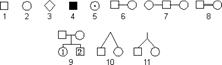
Условные обозначения, принятые при составлении родословных:
1 — мужчина; 2 — женщина; 3 — пол не выяснен; 4 — обладатель изучаемого признака; 5 — гетерозиготный носитель изучаемого рецессивного гена; 6 — брак; 7 — брак мужчины с двумя женщинами; 8 — родственный брак; 9 — родители, дети и порядок их рождения; 10 — дизиготные близнецы; 11 — монозиготные близнецы.
Благодаря генеалогическому методу были определены типы наследования многих признаков у человека. Так, по аутосомно-доминантному типу наследуются полидактилия (увеличенное количество пальцев), возможность свертывать язык в трубочку, брахидактилия (короткопалость, обусловленная отсутствием двух фаланг на пальцах), веснушки, раннее облысение, сросшиеся пальцы, заячья губа, волчья пасть, катаракта глаз, хрупкость костей и многие другие. Альбинизм, рыжие волосы, подверженность полиомиелиту, сахарный диабет, врожденная глухота и другие признаки наследуются как аутосомно-рецессивные.
Использование генеалогического метода показало, что при родственном браке, по сравнению с неродственным, значительно возрастает вероятность появления уродств, мертворождений, ранней смертности в потомстве. В родственных браках рецессивные гены чаще переходят в гомозиготное состояние, в результате развиваются те или иные аномалии. Примером этого является наследование гемофилии в царских домах Европы.
Близнецовый метод.
Это один из наиболее ранних методов изучения генетики человека, однако он не утратил своего значения и в настоящее время. Близнецовый метод был введен Ф.Гамильтоном, который выделил среди близнецов две группы:
•одняйцевые (монозиготные)
•двуяйцевые (дизиготные)
Монозиготные близнецы при нормальном эмбриональном развитии всегда одного пола. Дизиготные близнецы рождаются чаще (2/3 общего количества двоен), они развиваются из двух одновременно созревших и оплодотворенных яйцеклеток. Такие близнецы могут быть и однополые и разнополые. С генетической точки зрения они сходны как обычные сибсы, но у них большая общность факторов среды во внутриутробном (пренатальном) и частично в постнатальном периодах.
Если изучаемый признак проявляется у обоих близнецов пары, их называют конкордантными. Конкордантность – это процент сходства по изучаемому признаку. Отсутствие признака у одного из близнецов – дискордантность.
Близнецовый метод используется в генетике человека для того, чтобы оценить степень влияния наследственности и среды на развитие какого-либо нормального или патологического признака.
Методдерматоглифики
Дерматоглифка – это изучение рельефа кожи на пальцах, ладонях и подошвенных поверхностях стоп, который образован эпидермальными выступами – гребнями, которые образуют сложные узоры.
Ф. Гальтон предложил предложил классификацию этих узоров, позволившую использовать этот метод для идентификации личности в криминалистике.
Разделы дерматоглифики:
•дактилоскопия – изучение узоров на подушечках пальцев
•пальмоскопия – изучение рисунка на ладонях
•плантоскопия – изучение дерматоглифики подошвенной поверхности стопы
Дактилоскопия. Гребни на коже пальцев рук соответствуют сосочкам дермы, поэтому их называют также папиллярными линиями, рельеф этих выступов повторяет пласт эпидермиса. Межсосочковые углубления образуют бороздки. Закладка узоров происходит между 10 и 19 неделями внутриутробного развития; у 20 недельных плодов уже хорошо различимы формы узоров. Формирование папиллярного рельефа зависит от характера ветвления нервных волокон. Полное формирование деталей строения тактильных узоров отмечается к шести месяцам, после чего они остаются неизменными до конца жизни. Дерматоглифические исследования имеют важное значение в определении зиготности близнецов, в диагностике некоторых наследственных заболеваний, в судебной медицине, в криминалистике для идентификации личности.
Пальмоскопия. Ладонный рельеф очень сложный, в нем выделяют ряд полей, подушечек и ладонных линий. У правшей более сложные узоры встречаются на правой руке, у левшей – на левой. Индивидуальные особенности кожных узоров наследственно обусловлены. Это доказано многими генетическими исследованиями, в частности, на монозиготных близнецах.
Обширные исследования по изучению особенностей дерматоглифики проведены у нас в стране Т.Д. Гладковой (1996), а по наследственной обусловленности кожных узоров – И.С. Гусевой (1970, 1980). на основании этих работ был сделан вывод, что количественные показатели рельефа гребневой кожи программируются полигенной системой, включающей небольшое число аддитивно действующих генов. гены гребневой кожи проявляют свой морфогенетический эффект, влияя на степень ветвления нервного волокна, и фенотипически определяют гребневую плотность. На формирование дерматоглифических узоров могут оказывать влияние некоторые повреждающие факторы на ранних стадиях эмбрионального развития.
106.Методы медецинской генетики(популяционно-статистический, ДНК-анализ)
Популяц-статистич.
Одним из важных направлений в современной генетике является популяционная генетика. Она изучает генетическую структуру популяций, их генофонд, взаимодействие факторов, обусловливающих постоянство и изменение генетической структуры популяций. Под популяцией в генетике понимается совокупность свободно скрещивающихся особей одного вида, занимающих определенный ареал и обладающих общим генофондом в ряду поколений. (Генофонд — это вся совокупность генов, встречающихся у особей данной популяции).
В медицинской генетике популяционно-статистический метод используется при изучении наследственных болезней населения, частоты нормальных и патологических генов, генотипов и фенотипов в популяциях различных местностей, стран и городов. Кроме того, этот метод изучаетзакономерности распространения наследственных болезней в разных по строению популяциях и возможность прогнозировать их частоту в последующих поколениях.
Популяционно-статистический метод используется для изучения:
а) частоты генов в популяции, включая частоту наследственных болезней;
б) закономерности мутационного процесса;
ДНК-диагностика
Наиболее адекватные методы, обеспечивающие точную диагностику моногенных заболеваний, основаны на исследовании ДНК в районе определенных генов. Несмотря на то, что молекулярно-генетические методы, как правило, весьма сложны, трудоемки и дорогостоящи, данные, полученные в процессе ДНК-диагностики намного точнее и информативнее данных других анализов. Известно, что ДНК остается неизменной на протяжении всей жизни организма и одинакова во всех ядерных клетках, что позволяет использовать для анализа практически любые клетки организма, полученные на разных стадиях онтогенеза.
Прямые методы ДНК-диагностики используются в тех случаях, когда известен ген, ответственный за возникновение наследственного заболевания и основные типы его патологических мутаций. Использование прямых методов ДНК-диагностики целесообразно для таких заболеваний как муковисцидоз (мажорная мутация delF508), фенилкетонурия (R408W), хорея Гентингтона (экспансия CTG-повторов) и ряда других.
Главное преимущество прямого метода - это высокая, практически 100%, точность диагностики и отсутствие необходимости ДНК-анализа всех членов ядерной семьи. Обнаружение мутации в соответствующем гене позволяет абсолютно точно подтвердить диагноз наследственного заболевания и определить генотип всех членов отягощенной семьи. Еще одно достоинство прямой диагностики - возможность выявления гетерозиготного носительства патологических мутаций у родителей умершего больного и его родственников, что особенно актуально для аутосомно-рецессивных заболеваний.
Косвенные методы ДНК-диагностики применяют в том случае, если ген, повреждение в котором приводит к заболеванию, не идентифицирован, а лишь локализован на определенной хромосоме, или когда методы прямой ДНК-диагностики не дают результата, (например, при значительной протяженности и сложной молекулярной организации гена, а также широком спектре патологических мутаций в нем). Косвенные методы ДНК-диагностики основаны на анализе сегрегации в семье аллелей полиморфных маркеров, находящихся в том же хромосомном регионе или тесно сцепленных с локусом заболевания.
107.Методы профилактики наследственных болезней (медико-генетич консультирование, ген.скриинг,пренатальная диагностика)
Цель профилактического направления медицинской генетики — предотвратить или снизить риск возникновения заболеваний: • вновь появившихся вследствие мутации в половых клетках родителей; • унаследованных от предыдущих поколений; • возникающих в связи с наследственной предрасположенностью под влиянием факторов среды.
Медико-генетическое консультирование Медико-генетическое консультирование является основным видом профилактики врождённой и наследственной патологии.
Задача консультирования: сформулировать прогноз для потомства, течения заболевания, качества жизни и здоровья.
Пренатальная диагностика Пренатальная диагностика осуществляется в I и II триместрах беременности (в периоды, когда возможно прерывание беременности при обнаружении патологии плода).
Методы диагностики
• Ультразвуковое исследование (УЗИ) позволяет установить наличие беременности, количество плодов, выявлять грубые аномалии плода.
• Биохимическое исследование сыворотки крови матери: определение концентрации альфа-фетопротеина, хорионического гонадотропина, несвязанного эстриола и других веществ с целью диагностики ВПР и скрининга хромосомной патологии
• Фетоскопия обеспечивает прямое наблюдение плода с помощью специальной оптической системы, позволяет диагностировать заболевания кожи, нарушения развития половых органов, дефекты лица, конечностей и пальцев, производить биопсию тканей плода.
• Цитогенетические, биохимические и молекулярно-генетические методы применяют на материале, полученном при следующих вмешательствах:
- Амниоцентез позволяет получить амниотическую жидкость путём пункции амниона, проводят на 15—17-й неделе беременности.
- Кордоцентез обеспечивает возможность взятия крови плода путём пункции пуповины, проводят на 15—17-й неделе беременности
. - Хорионбиопсия — взятие для исследования ворсин хориона, клетки которого генетически идентичны клеткам развивающегося организма, проводят на 6—11-й неделе беременности.
108.Генетические основы профилактики наследственной патологии(первичная,втроричная,третичная)
Широкое распространение планирования семьи в развитых странах делает чрезвычайно важным вопрос об исходе каждой беременности. В связи с этим профилактика наследственных болезней должна занимать ведущее место в системе здравоохранения. Различают следующие виды профилактики наследственной патологии: первичная, вторичная и третичная профилактика.
Под первичной профилактикой понимают такие действия, которые должны предупредить рождение больного ребенка. Это реализуется через планирование деторождения путем выбора оптимального репродуктивного возраста, который для женщин составляет 21-35 лет (более ранние и поздние беременности увеличивают вероятность рождения ребенка с врожденной патологией и хромосомными болезнями), и отказа от деторождения в случаях высокого риска наследственной и врожденной патологии (в том числе при браках с кровными родственниками и гетерозиготными носителями патологического гена). Около 20 % всех наследственных болезней в каждом поколении - болезни, обусловленные новыми мутациями. В связи с этим важным элементом профилактики является жесткий контроль содержания мутагенов и тератогенов в окружающей среде.
Вторичная профилактика осуществляется путем прерывания беременности в случае высокой вероятности заболевания плода или пренатально диагностированной болезни. Прерывание производится только с согласия женщины и в установленные сроки. Прерывание беременности - решение явно не самое лучшее, к сожалению, в настоящее время оно является единственным практически пригодным при большинстве тяжелых и смертельных генетических дефектов.
Под третичной профилактикой наследственной патологии подразумевают коррекцию проявления патологических генотипов. С ее помощью можно добиться полной нормализации или снижения выраженности патологического процесса. Предотвращение развития наследственного заболевания включает в себя комплекс лечебных мероприятий, которые можно осуществлять внутриутробно или после рождения. В данном случае профилактические мероприятия тесно связаны с лечением наследственных болезней, и четкой границы между ними нет.
109. Основные принципы лечения наследственных болезней(этиологические,патогенетическое,симптоматическое лечение)
Симптоматическая терапия - это ликвидация тягостных для пациента проявлений заболевания, устранение отдельных симптомов болезни: кровоточивости, гиперестезии, подвижности зубов, клиновидных дефектов.
Симптоматическое лечение применяется практически при всех наследственных болезнях, даже тогда, когда врач располагает методами патогенетической терапии. Для многих форм наследственной патологии симптоматическое лечение - единственно возможное. Лекарственная симптоматическая терапия - наиболее часто используемый метод. Например, симптоматическая терапия миопатии включает в себя - витамины С, В, антихолинэстеразные препараты, АТФ, аминокислоты, анаболические гормоны и другие средства, способствующие улучшению метаболизма в мышцах. Успехи этого раздела терапии связаны с прогрессом фармакологии, обеспечивающим все более широкий выбор лекарств. Вместе с тем расшифровка патогенеза каждой болезни позволяет понять причину возникновения симптома, а на этой основе становиться возможной более тонкая лекарственная коррекция. Симптоматическое лечение включает в себя не только терапию лекарственными средствами. Многие физические методы лечения (климатотерапия, бальнеолечение, разные виды электротерапии, теплолечение) применяются при наследственных болезнях нервной системы, обмена веществ, заболеваниях скелета. У больных после курсов такой терапии улучшается самочувствие, увеличивается продолжительность жизни. Практически при всех наследственных болезнях показано физиотерапевтическое лечение. Например, лекарственная терапия муковисцидозе постоянно дополняется многообразными физиотерапевтическими процедурами (ингаляции, массаж и другие). К симптоматическому можно отнести рентгенорадиологическое лечение при наследственно обусловленных опухолях. Симптоматическое лечение (особенно лекарственное и диетическое) широко применяется и будет использоваться в будущем наряду с самым совершенным патогенетическим и этиотропным лечением наследственных болезней.
Этиологическое лечение. Наиболее перспективным и эффективным способом лечения наследственной патологии человека является коррекция генетического дефекта на уровне генов, то есть воздействие на этиологические факторы возникновения заболевания. Именно это направление в лечении наследственной патологии часто обозначают как генотерапия («молекулярное протезирование»). Разрабатываются три основные подхода к коррекции генетических дефектов посредством гемотерапии: I) компенсация экспрессии функционально неактивных аллелей введением в клетку дополнительных копий гена; 2) угнетение избыточной экспрессии гена; 3) усиление иммунного ответа организма. Первый подход основан на введении в определенные клетки и ткани организма дополнительного генетического материала, корригирующего нарушение экспрессии одного или нескольких мутантных генов. Этот подход наиболее часто используют дли коррекции генетического дефекта при моногенных и некоторых мультифакториальных заболеваниях. Коррекция генетического дефекта осуществлется как в половых, так и в соматических клетках организма. Более предпочтительна генотерапия на уровне соматических клеток, так как она позволяет модифицировать экспрессию генов в определенном типе клеток и тканей и не приводит к передаче измененной генетической информации в ряду поколений. Второй подход основан на подавлении избыточной функции генов и их продуктов в клетках. Этот подход чрезвычайно перспективен для лечения онкологических заболеваний. В этом случае используют несколько генотерапевтических методов: 1) введение генов, продукты которых приводят к гибели избыточно пролиферируюших клеток (генов-убииц); 2) блокирование экспрессии онкогенов путем введения антисмысловых нуклеотидных последовательностей или генов, продукты экспрессии которых являются антителами для ряда белковых продуктов опухолевой клетки; 3) введение в опухолевые клетки нормальных копий генов-супрессоров. Третий подход направлен на повышение иммунореактивности клеток-мишеней или активации иммунной системы организма и разрабатывается для онкологических и вирусных заболеваний.
Патогенетическое лечение направлено на коррекцию биохимических и физиологических процессов, нарушенных в результате изменения концентрации белкового продукта мутантного гена. Этот метод лечения наиболее эффективен при наследственных болезнях обмена, основным патогенетическим механизмом которых является нарушение утилизации субстрата. Воздействие на процессы обменных превращений может осуществляться несколькими путями и зависит, прежде всего, от того являются ли патологические симптомы заболевания следствием нарушения утилизации субстрата (вводимого извне или синтезированного в организме) или они обусловлены недостатком продуктов его метаболизма в организме больного
В целом патогенетические подходы к лечению наследственных болезней систематизирован но в зависимости от уровня биохимического дефекта можно представить следующим образом. Суть любых подходов к лечению схематично сводится к возмещению или выведению чего-то. Если ген не работает, то необходимо возместить его продукт; если ген производит не то, что нужно, и образуются токсичные продукты, то необходимо удаление таких продуктов и возмещение основной функции; если ген производит много продукта, то избыток последнего удаляют.

