




Билет №15
Витамин С.
Аскорби́новая кислота́ — органическое соединение, родственное глюкозе, является одним из основных веществ в человеческом рационе, которое необходимо для нормального функционирования соединительной и костной ткани. Выполняет биологические функции восстановителя и кофермента некоторых метаболических процессов, является антиоксидантом. Биологически активен только один из изомеров — L-аскорбиновая кислота, который называют витамином C. В природе аскорбиновая кислота содержится во многих фруктахи овощах. По физическим свойствам аскорбиновая кислота представляет собой белый кристаллический порошок кислого вкуса. Легко растворим в воде, растворим в спирте. Биологическая роль. Витамин С (аскорбиновая кислота) повышает защитные силы организма, ограничивает возможность заболеваний дыхательных путей, улучшает эластичность сосудов (нормализует проницаемость капилляров). Витамин оказывает благоприятное действие на функции центральной нервной системы, стимулирует деятельность эндокринных желез, способствует лучшему усвоению железа и нормальному кроветворению, препятствует образованию канцерогенов. Большие дозы полезны для больных сахарным диабетом, заядлых курильщиков, женщин, пользующихся противозачаточными препаратами, для пожилых людей с пониженной способностью пищеварительного тракта всасывать витамины.
Недостаток проявляется в быстрой утомляемости, кровоточивости десен, в общем снижении устойчивости организма против инфекций, при далеко зашедшем гиповитаминозе С может появится цинга, для которой характерны разрыхление, опухание и кровоточивость десен и выпадение зубов, мелкие подкожные кровоизлияния. При передозировке возможны нарушения функции печени и поджелудочной железы. Образование коллагена, серотонина из триптофана, образование катехоламинов, синтез кортикостероидов. Аскорбиновая кислота также участвует в превращении холестерина в желчные кислоты. Витамин С необходим для детоксикации в гепатоцитах при участии цитохрома P450. Витамин С сам нейтрализует супероксид-анион радикал до перекиси водорода. Восстанавливает убихинон и витамин Е. Стимулирует синтез интерферона, следовательно, участвует в иммуномодулировании. Переводит трёхвалентное железо в двухвалентное, тем самым способствует его всасыванию. Тормозит гликозилирование гемоглобина, тормозит превращение глюкозы в сорбит.
Содержится в свежих растениях: шиповнике, кизиле, черной смородине, рябине, облепихе, цитрусовых плодах, красном перце, хрене, петрушке, зеленом луке,укропе, кресс-салате, краснокачанной капусте, картофеле, брюкве, капусте, в овощной ботве. В лекарственных растениях: крапиве, будре, любистоке, в лесных плодах.
Строение витамина С было окончательно установлено синтезом его из L-кислоты. Витамин С получил название L-аскорбиновой кислоты. L-Аскорбиновая кислота представляет собой кристаллическое соединение, легко растворимое в воде с образованием кислых растворов. Наиболее замечательной особенностью этого соединения является его способность к обратному окислению (дегидрированию) с образованием дегидроаскорбиновой кислоты. Таким образом, L-аскорбиновая кислота и её дегидроформа образуют окислительно-восстановительную систему, которая может как отдавать, так и принимать водородные атомы, точнее электроны и протоны. Обе эти формы обладают антискорбутным действием. В присутствии широко распространённого в растительных тканях фермента - аскорбиноксидазы, или аскорбиназы, аскорбиновая кислота окисляется кислородом воздуха с образованием дегидроаскорбиновой кислоты и перекиси водорода. Аскорбиновая кислота, особенно её дегидроформа, является весьма неустойчивым соединением. Превращение в дикетоулоновую кислоту, не обладающую витаминной активностью, является необратимым процессом, который заканчивается обычно окислительным распадом. Наиболее быстро витамин С разрушается в присутствии окислителей в нейтральной или щелочной среде при нагревании. Поэтому при различных видах кулинарной обработки пищи часть витамина С обычно теряется, аскорбиновая кислота обычно разрушается также и при изготовлении овощных и фруктовых консервов. Особенно быстро витамин С разрушается в присутствии следов солей, тяжёлых металлов (железо, медь). В настоящее время, однако, разработаны способы приготовления консервированных фруктов и овощей с сохранением их полной витаминной активности.
Отдавая два атома водорода, аскорбиновая кислота окисляется в дегидроаскорбиновую кислоту. Реакция эта обратима: дегидроаскорбиновая кислота, присоединяя два атома водорода, легко восстанавливается в аскорбиновую кислоту. Этот водород дегидроаскорбиновая кислота может получать от восстановленной формы кофермента дегидрогеназы. А послед-ний его приобретает, отнимая от различных субстратов, окисляющихся путем отщепления водорода. Таким образом, система аскорбиновая кислота -- дегидроаскорбиновая кислота принимает участие в транспорте водорода (электронов и протонов), то есть в реакциях окисления -- восстановления некоторых продуктов обмена веществ.
Важную роль в этих реакциях играет свободный радикал монодегидроаскорбиновой кислоты -- продукт отщепления не двух, а одного электрона от аскорбиновой кислоты.
Окисление аскорбиновой кислоты катализируется в растениях специфическим ферментом аскорбатоксидазой, содержащим в своей молекуле белок, связанный с медью. Сама медь и различные ее соединения также являются мощными, хотя и неспецифическими катализаторами окисления аскорбиновой кислоты. Важную роль в окислении витамина С в животном организме, где нет абкорбатоксидазы, играет медьсодержащий белок церулоплазмин.
Дегидроаскорбиновая кислота, являющаяся первым продуктом в цепи реакций распада аскорбиновой кислоты, очень неустойчивое соединение. Судьба ее может быть двоякой. Если среда, в которой находится дегидроаскорбиновая кислота, способствует протеканию восстановительной реакции, она присоединяет к себе водород, превра-щаясь в исходный продукт -- аскорбиновую кислоту. Поэтому нередко дегидроаскорбиновую кислоту называют обратимо окисленной формой витамина С. Если же условия среды не благоприятствуют восстановлению, она подвергается необратимому распаду, окисляясь до продуктов, которые уже не превращаются в аскорбиновую кислоту.
Как установили В. А. Энгельгардт и В. Н. Букин, распад дегидроаскорбиновой кислоты не требует наличия специальных ферментов, он происходит самопроизвольно. Среди условий, влияющих на скорость окисления аскорбиновой кислоты, кроме катализаторов, очень важную роль играет реакция среды: витамин С относительно более устойчив в кислой реакции среды, малоустойчив в нейтральной и чрезвычайно быстро распадается в щелочной. Существенное значение для устойчивости витамина С имеет присутствие в среде других веществ: одни из них (сахара, аминокислоты) благоприятствуют сохранности - аскорбиновой кислоты, другие (например, соединения меди) способствуют ее окислительному распаду. Чтобы закончить ознакомление с химической природой и свойствами витамина С, остается упомянуть о связанный формах аскорбиновой кислоты, так называемом аскорбигене. Это двухкомпонентные соединения, состоящие из аскорбиновой кислоты, связанной с белками, нуклеиновыми кислотами, производными азотистого соединения индола либо так называемым витамином Р. Биологическая роль связанных форм витамина С еще не выяснена. По этому поводу имеются различные гипотезы. Возможно, что это транспортные формы витамина, находясь в составе которых он защищен от окисления и разносится по организму. Не исключено, что в соединении с белком аскорбиновая кислота несет коферментную функцию в неизвестной еще ферментной системе. Природа и функции связанных форм витамина С находятся в процессе изучения.
2. Окисление пальмитиновой кослоты.
Процесс окисления жирной кислоты в митохондриях клетки включает несколько последовательных энзиматических реакций.
Первая стадия дегидрирования. Ацил-КоА в митохондриях прежде всего подвергается ферментативному дегидрированию, при этом ацил-КоА теряет 2 атома водорода в α- и β-положениях, превращаясь в КоА-эфир ненасыщенной кислоты. Таким образом, первой реакцией в каждом цикле распада ацил-КоА является его окисление ацил-КоА-де-гидрогеназой, приводящее к образованию еноил-КоА с двойной связью между С-2 и С-3:

Существует несколько ФАД-содержащих ацил-КоА-дегидрогеназ, каждая из которых обладает специфичностью по отношению к ацил-КоА с определенной длиной углеродной цепи.
Стадия гидратации. Ненасыщенный ацил-КоА (еноил-КоА) при участии фермента еноил-КоА-гидратазы присоединяет молекулу воды. В результате образуется β-оксиацил-КоА (или 3-гидроксиацил-КоА):

Заметим, что гидратация еноил-КоА стереоспецифична, подобно гидратации фумарата и аконитата. В результате гидратации транс-Δ2-двойной связи образуется только L-изомер 3-гидроксиацил-КоА.
Вторая стадия дегидрирования. Образовавшийся β-оксиацил-КоА (3-гидроксиацил-КоА) затем дегидрируется. Эту реакцию катализируют НАД+-зависимые дегидрогеназы:
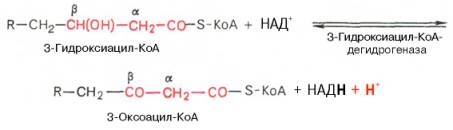
Тиолазная реакция. В ходе предыдущих реакций происходило окисление метиленовой группы при С-3 в оксогруппу. Тиолазная реакцияпредставляет собой расщепление 3-оксоацил-КоА с помощью тиоловой группы второй молекулы КоА. В результате образуется укороченный на два углеродных атома ацил-КоА и двууглеродный фрагмент в виде ацетил-КоА. Данная реакция катализируется ацетил-КоА-ацилтрансферазой (β-ке-тотиолазой):

Образовавшийся ацетил-КоА подвергается окислению в цикле трикар-боновых кислот, а ацил-КоА, укоротившийся на два углеродных атома, снова многократно проходит весь путь β-окисления вплоть до образования бутирил-КоА (4-углеродное соединение), который в свою очередь окисляется до 2 молекул ацетил-КоА (рис. 11.2). Например, при окислении пальмитиновой кислоты (С16) повторяется 7 циклов β-окисления. Запомним, что приокислении жирной кислоты, содержащей п углеродных атомов, происходит n/2–1 цикл β-окисления (т.е. на один цикл меньше, чем n/2, так как при окислении бутирил-КоА сразу происходит образование 2молекул ацетил-КоА) и всего получится п/2 молекул ацетил-КоА. Следовательно, суммарное уравнение β-окисления активированной кислоты можно записать так:
Пальмитоил-КоА + 7ФАД + 7НАД+ + 7Н2O + 7HS-KoA –>
–> 8Ацетил-КоА + 7ФАДН2 + 7НАДН + 7Н+.
Баланс энергии. При каждом цикле β-окисления образуются одна молекула ФАДН2 и одна молекула НАДН. Последние в процессе окисления вдыхательной цепи и сопряженного с ним фосфорилирования дают: ФАДН2 – 2 молекулы АТФ и НАДН – 3 молекулы АТФ, т.е. в сумме за один цикл образуется 5 молекул АТФ. При окислении пальмитиновой кислоты образуется 5 х 7 = 35 молекул АТФ. В процессе β-окисления пальмитиновой кислоты образуется 8 молекул ацетил-КоА, каждая из которых, «сгорая» в цикле трикарбоновых кислот, дает 12 молекул АТФ, а 8 молекул ацетил-КоА дадут 12 х 8 = 96 молекул АТФ.
Таким образом, всего при полном β-окислении пальмитиновой кислоты образуется 35 + 96 = 131 молекула АТФ. С учетом одной молекулы АТФ, потраченной в самом начале на образование активной формы пальмитиновой кислоты (пальмитоил-КоА), общий энергетический выход при полномокислении одной молекулы пальмитиновой кислоты в условиях животного организма составит 131 – 1 = 130 молекул АТФ. Изменение свободной энергии ΔF при полном сгорании 1 моля пальмитиновой кислоты составляет 2338 ккал, а богатая энергией фосфатная связь АТФ характеризуется величиной 7,6 ккал/моль. Нетрудно подсчитать, что примерно 990 ккал (7,6 х 130), или 42% от всей потенциальной энергии пальмитиновой кислотыпри ее окислении в организме, используется для ресинтеза АТФ, а оставшаяся часть, очевидно, теряется в виде тепла.
Следовательно, эффективность накопления энергии в результате окисления жирных кислот при стандартных условиях составляет ~ 40%, что близко к соответствующей величине для гликолиза, цикла трикарбоновых кислот и окислительного фосфорилирования.
3.Биосинтез мочевины.
Мочевина является главным конечным продуктом обмена аминокислот. Синтезируется мочевина из аммиака, который постоянно образуется в организме при окислительном и неокислительном дезаминировании аминокислот, при гидролизе амидов глутаминовой и аспарагиновой кислот, а также при распаде пуриновых и пиримидиновых нуклеотидов. Часть аммиака образуется в кишечнике в результате действия бактерий на пищевые белки (гниение белков в кишечнике) и поступает в кровь воротной вены. Аммиак - токсичное соединение. Даже небольшое повышение его концентрации оказывает неблагоприятное действие на организм, и прежде всего - на центральную нервную систему. Несмотря на то, что аммиак постоянно продуцируется в тканях, он содержится в периферической крови лишь в следовых количествах, так как быстро удаляется из кровеносной системы печенью, где входит в состав глутамата, глутамина и мочевины. Биосинтез мочевины является основным механизмом обезвреживания аммиака в организме. Синтез мочевины происходит в печени в цикле Кребса-Гензелейта (другое название - орнитиновый цикл мочевинообразования Кребса) в несколько этапов с участием ряда ферментных систем. Синтез сопровождается поглощением энергии, источником которой является АТФ.
Весь цикл мочевинообразования можно представить следующим образом:
На первом этапе синтезируется карбамоилфосфат в результате конденсации ионов аммония, двуокиси углерода и фосфата (поступающего из АТФ) под действием фермента карбамоилсинтетазы. Карбамоилфосфат - это метаболически активная форма аммиака, используемая в качестве исходного продукта для синтеза ряда других азотистых соединений.
На втором этапе мочевинообразования происходит конденсация карбамоилфосфата и орнитина с образованием цитруллина; реакцию катализирует орнитинкарбамоилтрансфераза.
На следующей стадии цитруллин превращается в аргинин в результате двух последовательно протекающих реакций. Первая из них, энергозависимая, сводится к конденсации цитруллина и аспарагиновой кислоты с образованием аргининосукцината (эту реакцию катализирует аргининосукцинатсинтетаза). Аргининосукцинат распадается в следующей реакции на аргинин и фумарат при участии другого фермента - аргининосукцинатлиазы.
На последнем этапе аргинин расщепляется на мочевину и орнитин под действием аргиназы.
Эффективность работы орнитинового цикла при нормальном питании человека и умеренных физических нагрузках составляет примерно 60% его мощности. Запас мощности необходим для избежания гипераммониемии при изменении количества белка в пище. Увеличение скорости синтеза мочевины происходит при длительной физической работе или длительном голодании, которое сопровождается распадом тканевых белков. Некоторые патологические состояния, характеризующиеся интенсивным распадом белков тканей (сахарный диабет и др.) также сопровождаются активацией орнитинового цикла.
Нормальный ход метаболического превращения аммиака в мочевину имеет большое значение для организма. При серьезных нарушениях функции печени - например, при обширном циррозе или тяжелом гепатите - аммиак, являясь токсичным веществом, накапливается в крови, вызывая тяжелые клинические симптомы. Известны врожденные метаболические нарушения, связанные с недостатком одного из ферментов, участвующих в синтезе мочевины. Все нарушения синтеза мочевины вызывают аммиачное отравление.
Синтезированная в печени мочевина попадает в кровь, затем в почки и в итоге выводится с мочой. Мочевина является беспороговым веществом: все образующееся количество фильтруется в просвет проксимальных канальцев, а затем часть (около 35 %) реабсорбируется обратно за счет реабсорбции воды. В связи с этим величина экскреции мочевины является менее информативным показателем клубочковой фильтрации, чем показатель, основывающийся на экскреции креатинина (который, в отличие от мочевины, практически не реабсорбируется).

Рис. 12.5. Орнитиновый цикл синтеза мочевины в печени.
Аммиак образуется главным образом в процессе глутаматде-гидрогеназной реакции. В процессе пополнения запасов аспартата участвуют три сопряженные реакции: сначала фумарат под действием фумаразы присоединяет воду и превращается в малат, который окисляется при участии малатдегидрогеназы с образованием оксалоацетата; последний в реакции трансаминирования с глутаматом вновь образует аспартат.
Учитывая известные фактические данные о механизмах обезвреживания аммиака в организме, можно сделать следующее заключение. Часть аммиака используется на биосинтез аминокислот путем восстановительного аминирования α-кетокислот по механизму реакции трансаминирования. Аммиак связывается при биосинтезе глутамина и аспарагина. Некоторое количество аммиака выводится с мочой в виде аммонийных солей. В форме креатинина, который образуется из креатина и креатинфосфата, выделяется из организма значительная часть азота аминокислот.
Билет №16
1. Витамин А.
Каротиноиды объединяют группу производных растительных пигментов каротинов. Наибольшее значение имеет ретинол (А1) и дегидроретинол (А2). Среди биологически активных соединений важнейшими считаются α-, β- и γ-каротины. Наибольшую ценность для организма представляет β-каротин, который содержит 2 β-ионовых кольца, соединенных цепью, состоящей из 18 атомов углерода (из 4 частиц изопрена). Этот каротин широко распространен в природе.
Молекула витамина А содержит только одно β-ионовое кольцо, а боковая цепь состоит из 2 частей изопрена. Молекула витамина А представляет собой половину молекулы β-каротина, который является провитамином витамина А. Провитамином обычно называют непосредственный предшественник, из которого образуется витамин. Поскольку в молекуле витамина А есть гидроксильная группа, он является высокомолекулярным циклическим одноатомным ненасыщенным первичным спиртом. Превращение β-каротина в витамин А происходит преимущественно в стенке тонкой кишки, а также в печени. В этих органах есть и специфический фермент, катализирующий гидролитический распад β-каротина на 2 молекулы А-альдегида-15-15׳-каротин-диоксигеназа. В 1937 году из печени пресноводных рыб был выделен витамин, из которого в цикле на 2 атома водорода меньше, чем у витамина А1. Его назвали витамином А2, дегидроретинолом. А1 и А2 обладают одинаковым биологическим действием и физико-химическими свойствами, только витамин А2 менее активен. Оба витамина получены в чистом виде и синтезированы.

Они хорошо растворяются в жирах и жировых растворителях, достаточно устойчивы к действию щелочей. Витамин А термостабилен, может выдерживать стерилизацию без доступа кислорода. На воздухе он быстро окисляется и разрушается, особенно в кислой среде. Этому способствует также солнечное освещение.. Биологически активными формами витамина А в организме человека и животных могут быть витамин А-спирт, витамин А-альдегид, витамин А-кислота, а также эфиры витамина А. В кишки витамин А попадает в виде эфира, все другие формы образуются уже в тканях. Каждая из этих структур играет определенную роль в обмене веществ, формировании структуры и функциях клеток. Например, витамин А-спирт (в виде эфиров с жирными кислотами) является основным резервом витамина А в тканях, витамин А-альдегид нужен для образования зрительных пигментов, а витамин А-кислота – для нормального роста животных и некоторых других процессов.
Одной из важных функций витамина А является его участие в образовании сложного белка родопсина (зрительного пурпура) сетчатки глаза. Родопсин, имеющийся в палочках, и являющийся фоточувствительным пигментом, состоит из белка опсина и альдегидной формы витамина А – ретиналя. Ретиналь образуется отщеплением 2 атомов водорода от первичной спиртовой группы витамина; он может находиться в цис- и транс-формах. Под действием света цис-ретиналь переходит в транс-ретиналь, после чего родопсин распадается на белок опсин и ретиналь. В темноте эти части снова соединяются, благодаря чему создается возможность видеть в сумерках и ночью. При отщеплении ретинола от родопсина часть его разрушается, поэтому для ресинтеза молекулы родопсина нужны новые молекулы витамина А. Если их нет, то образование ретинола, а в связи с этим и родопсина, затормаживается. В результате этого человек теряет способность видеть в сумерках, т.е. развивается «куриная слепота». Витамин необходим для синтеза нуклеиновых кислот и белков, в частности, белков сыворотки крови, для нормального обмена липидов (в митохондриях печени при А-гиповитаминозе обнаружено уменьшение содержания общих липидов и фосфолипидов, непредельных жирных кислот (арахидоновой и линоленовой) при одновременном увеличении содержания холестерина и триглицеридов). Витамин А влияет на активность ферментов тканевого дыхания и на процессы окислительного фосфорилирования, а также на обмен минеральных веществ, в частности, солей кальция. Каротины в иммунной системе повышают защитную силу собственных интерферонов организма против возбудителей болезней. Прежде всего они защищают от свободных радикалов вилочковую железу, которая представляет собой штаб-квартиру иммунной системы.
Витамин А обнаружен только в организме человека и животных. В растениях содержатся его провитамины – каротины. Всасывание витамина А в кишках происходит при участии желчных кислот. Все факторы, нарушающие переваривание и всасывание жиров, задерживают всасывание каротина и витамина А. Витамин А переносится кровью в комплексе с белком-переносчиком, т.н. ретинол-связывающим протеином (РСП). При недостатке белка в диете снижается депонирование витамина А в печени и образование его из β-каротина. Основным депо витамина А является печень, где он откладывается в виде белково-витаминных комплексов. В печени же основная масса β-каротина превращается в витамин А. Важнейшие источники витамина А: печень, сливочное масло, сливки, сыр, яичный желток, рыбий жир.
2. Окисление ненасыщенных жирных кислот.
Ненасыщенные жирные кислоты, как и насыщенные, подвергаются β-окисление. Положение и число двойных связей в молекулах ненасыщенных жирных кислот определяют особенности их окисления. НЖК окисляются как насыщенные до места двойной связи. Если двойная связь имеет трансконфигурацию и расположение, как в еноил-КоА, образующемся при окислении насыщенных жирных кислот, то дальше окисление идет обычным путем. При отсутствии этого условия вступает в действие дополнительный фермент, перемещающий двойную связь и меняющий цис- в трансконфигурацию. Двойная связь может восстанавливаться НАДФ•Н2. Е. А. Строев (1986) отмечает, что скорость окисления ненасыщенных жирных кислот очень высока: олеиновой кислоты в 11 раз, линолевой — в 114, линоленовой — в 170, арахидоновой — в 200 раз выше, чем стеариновой. В исследованиях с олеиновой кислотой, меченной дейтерием, было установлено, что она может редуцироваться, превращаясь в стеариновую, а последняя подвергается β-окислению (И. В. Савицкий, 1973,1982). Такой путь допускали и для других ненасыщенных жирных кислот. Однако их окисление происходит иначе. На первом этапе под действием липоксигеназы происходит дегидрирование (отщепление) одного атома водорода и жирная кислота превращается на свободный радикал. Липоксигеназы широко представлены в тканях животных и обнаружены в микросомальной фракции гомогенатов клеток. Они катализируют реакции переокисления жирных кислот, которые различаются расположением окисляемого кислородом атома углерода. (В микросомальных мембранах образование перекисей катализирует диоксигеназа фосфолипидов, зависящая от НАДФ-Н2. Для образования перекисей липидов необходимо участие негеминового железа, которое в дальнейшем восстанавливается в микросомальной электронно-транспортной цепи). Образование под влиянием липоксигеназы свободного радикала обусловливает перестройку всей молекулы жирной кислоты. В результате этого превращения двойные связи из изолированных становятся сопряженными (приближаются одна к одной), а кислота с сопряженными двойными связями при наличии кислорода, по мнению автора, окисляется с образованием гидроперекиси и цикличной перекиси. Перекиси и гидроперекиси разлагаются до отдельных фрагментов — жирного альдегида (к примеру, капронового), малонового диальдегида, полуальдегида дикарбоновой кислоты. При этом имеется прямая зависимость количества образовавшегося малонового диальдегида от количества двойных связей в молекуле ненасыщенной жирной кислоты: линолевая образует одну молекулу малонового диальдегида, линоленовая — две, арахидоновая — три, клупанодоновая — четыре. В качестве примера можно привести окисление линолевой кислоты: она последовательно превращается в свободный радикал линолевой кислоты, затем в ненасыщенную кислоту с двойными сопряженными связями, дальше в гидроперекись и циклическую перекись линолевой кислоты, которые разлагаются на капроновый альдегид, малоновый диальдегид и полуальдегид азелаиновой кислоты. Последние три продукта расщепления претерпевают дальнейшее окисление: образуются капроновая, азелаиновая и малоновая кислоты. Капроновая кислота после превращения в капронилкоэнзим А подвергается β-окислению. Азелаиновая кислота также включается в β-окисление, а малоновая после декарбоксилирования превращается в уксусную кислоту. Таким образом, линолевая кислота превращается в остатки уксусной кислоты, которые затем в цикле Кребса окисляются до СО2 и Н2О. Аналогичным образом (но с другими промежуточными продуктами) окисляются и другие ненасыщенные жирные кислоты: при окислении линоленовой кислоты образуется пропионовая, азелаиновая и две молекулы малоновой кислоты, при окислении арахидоновой — капроновая, глютаровая и три молекулы малоновой кислоты. Путем многостадийного процесса линолевая кислота может сначала превратиться в арахидоновую, которая затем подвергается окислению. Таким образом, в данном случае ненасыщенные жирные кислоты подвергаются β-окислению, но это наступает на более поздних этапах после их предварительной фрагментации и образования альдегидов с короткой углеродной цепью. Однако следует напомнить, что приведенная выше в качестве примера окисления линолевая кислота используется для синтеза арахидоновой кислоты и в фосфолипидах тканей содержится лишь в следовых количествах.
Окисление ненасыщенных жирных кислот в принципе происходит так же, как и окисление насыщенных жирных кислот, но с некоторыми особенностями. Двойные связи природных ненасыщенных жирных кислот (олеиновой, линолевой и т.д.) имеют цис-конфигурацию, а в КоА-эфирах ненасыщенных кислот, являющихся промежуточными продуктами при β-окислении насыщенных жирных кислот, двойные связи имеют трансконфигурацию. Кроме того, последовательное удаление двууглеродных фрагментов при окислении ненасыщенных жирных кислот до первойдвойной связи дает Δ3,4-ацил-КоА, а не Δ2,3-ацил-КоА, который является промежуточным продуктом при β-окислении ненасыщенных жирных кислот:

В тканях существует фермент, который осуществляет перемещение двойной связи из положения 3–4 в положение 2–3, а также изменяет конфигурацию двойной связи из цис- в транс-положение. Этот фермент получил название Δ3,4-цис –> Δ2,3-транс-еноил-КоА-изомеразы.
Например, арахидоновая кислота может образоваться в клетке только при наличии линоленовой или линолевой кислот. При этом линолевая кислота (18:2) дегидрируется до γ-линоленовой (18:3) и удлиняется до эйкозотриеновой кислоты (20:3), последняя далее вновь дегидрируется до арахидоновой кислоты (20:4). Так формируются жирные кислоты ω6 ряда
Для образования жирных кислот ω3-ряда, например, тимнодоновой (20:5), необходимо наличие α-линоленовой кислоты (18:3), которая дегидрируется (18:4), удлиняется (20:4) и опять дегидрируется (20:5).
3. Превращение безазотистого скелета ак
В состав белков входит 20 обычных аминокислот, различающимися своими углеродными скелетами. Соответственно, существует и 20 различных катаболических путей для их расщепления. Из общего количества энергии, потребляемой организмом, на долю всех этих путей приходиться не более 10%. Значение этих аминокислотных путей, взятых по отдельности, не может идти ни в какое сравнение со значением гликолиза или цикла трикарбоновых кислот. Трансаминирование и дезаминирование аминокислот ведет к образованию безазотистых углеродных скелетов аминокислот – α-кетокислот. В состав белков входят 20 аминокислот, различающихся по строению углеводородного радикала, каждый из которых катаболизируется по своим специфическим метаболическим путям, окисляясь полностью до СО2 и Н2О.
Катаболизм всех аминокислот сводится к образованию шести веществ, вступающих в общий путь катаболизма: пируват, ацетил-КоА, a-кетоглутарат, сукцинил-КоА, фумарат, оксалоацетат. 5 аминокислот превращаются в £-кетоглутарат, 3 – в сукцинил-КоА, 2 – в оксалоацетат и 2 – в фумарат. Углеродные скелеты 10 аминокислот, разрушаясь, превращаются в ацетил-КоА, непосредственно включающийся в ЦТК. 5 из 10 аминокислот расщепляются до ацетил-КоА через пируват; другие 5 превращаются сначала в ацетоацетил-КоА, а потом расщепляется до ацетил-КоА. Через пируват идет расщепление аланина, цистеина, глицина, серина и треонина. Аланин превращается в пируват непосредственно в реакции трансаминирования с £-кетоглутаратом. Треонин расщепляется с образованием глицина, который превращается в серин (в результате ферметативного присоединения гидроксиметильной группы) либо окисляется СО2, NН4 и метиленовой группы. Фрагменты углеродного скелета енилаланина, тирозина, лизина, триптофана и лейцина превращаются в ацетоацетил-КоА, из которого потом образуется ацетил-КоА. Из фенилаланина и тирозина образуется по 2 4-углеродных продукта – ацетоацетат и фумарат. Ацетоацетат поступает в ЦТК в форме ацил-КоА, а фумарат сам является промежуточным продуктом этого цикла. Углеродные скелеты аспарагина и аспартата поступают в ЦТК через оксалоацетат. Фермент аспарагиназа катализирует гидролиз аспарагина с образование аспартата. Аминогруппа аспартата передается затем £-кетоглутарату в реакции трансаминирования, продуктом которой является глутамат. Остающийся углеродный скелет аспартата, в форме оксалоацетата, включается в ЦТК. Аминокислоты, которые превращаются в промежуточные продукты ЦТК (a-кетоглутарат, сукцинил-КоА, фумарат), и образуют в конечном итоге оксалоацетат, могут использоваться в процессе глюконеогенеза. Такие аминокислоты называются гликогенными. К ним относятся: аланин, аргинин, аспартат, глутамат, глицин, гистидин, метионин, пролин, серин, треонин, валин, цистеин. Катаболизм лейцина и лизина не включает стадии образования пировиноградной кислоты, их углеводородная часть превращается непосредственно в ацетоацетат (лейцин, лизин) или в ацетил-КоА (лейцин) и используются в синтезе кетоновых тел.
Тирозин, фенилаланин, изолейцин и триптофан являются смешанными или одновременно гликогенными и кетогенными. Часть углеродных атомов их молекул при катаболизме образует пируват, другая часть включается в ацетил-КоА, минуя стадию пирувата. Истинной кетогенной аминокислотой является лейцин.
Билет №17
1.Витамин Д
Кальциферолы объединяют группу производных стеринов растительного и растительного происхождения, обладающих антирахитическим действием. Одним из наиболее распространенных и изученных провитаминов является эргостерин – производное циклопентанпергидрофенантрена. Другие провитамины отличаются от эргостерина только особенностями строения боковой цепи.

Эргостерин
Витамин D, образующийся при облучении ультрафиолетовыми лучами дрожжевого эргостерина, назвали витамином D2, или эргокальциферолом, а витамин D, который образуется из –дегидрохолестерина, - витамином D3, или холекальциферолом. Раньше неочищенный препарат кальциферола называли витамином D1. Витамин D2, полученный их дрожжей, относительно устойчив к действию высоких температур и окислению, он хорошо растворяется в жирах и органических растворителях. 
Витамин D3 образуется в организме человека и животных, особенно в коже, под действием солнечной и искусственной ультрафиолетовой радиации из 7-дегидрохолестерина.

Как видно из приведенной формулы, витамин D3 имеет на одну двойную связь больше, чем его провитамин. Эта связь образуется в результате разрыва кольца в 7-дегидрохолестерине в положениях 9 и 10. витамин D оказывает влияние на фосфорно-кальциевый обмен, от всасывания и распределения в тканях до выделения из организма продуктов обмена. В этой связи важное звено биологического действия кальциферола составляет его воздействие на формирование костной ткани. Установлено, что в печени витамин D3 превращается в свой активный метаболит – 25-гидроксихолекальциферол, антирахитическая активность которого в 1,4 раза выше, чем витамина D3. В почках 25-гидроксихолекальциферол подвергается дальнейшим превращениям с образованием 1,25-дигидроксихолекальциферола, обладающего в несколько раз более интенсивным действием на всасывание кальция в тонкой кишке и мобилизацию его в костной ткани, чем 25-гидроксихолекальциферол. Полагают, что формой, ответственной за антирахитическое воздействие витамина в организме, является не сам витамин D, а его гидроксилированные производные. Витамин D способствует всасыванию кальция и фосфора в кишках, обеспечивая перенос их через стенку кишок даже против градиента концентрации; влияет также на реадсорбцию фосфата в почках, способствуя эффективному использованию его в организме; способствует обратному всасыванию в почках некоторых аминокислот, особенно оксипролина – важной составной части коллагена; влияет на процессы тканевого дыхания, в частности, на окисление углеводов. Основным источником витамина D для человека в летнее время составляет собственный синтез витамина D (витамин D3) в коже под воздействием ультрафиолетового излучения. Достаточно 15-20 минут в день воздействия ультрафиолета солнечного излучения для удовлетворения потребности организма в витамине D. Основным источником витамина D являются продукты питания: сливочное масло, сыр и другие молочные продукты, яичный желток, рыбий жир, икра. Растительные источники - люцерна, хвощ, крапива, петрушка, грибы, семена подсолнечника. Наибольшее количество витамина D содержится в рыбьем жире.
2. Биосинтез высших жирных кислот.
В настоящее время в достаточной степени изучен механизм биосинтеза жирных кислот в организме животных и человека, а также катализирующие этот процесс ферментные системы. Синтез жирных кислот протекает в цитоплазме клетки. В митохондриях в основном происходит удлинение существующих цепей жирных кислот. Установлено, что в цитоплазме печеночных клеток синтезируется пальмитиновая кислота (16 углеродныхатомов), а в митохондриях этих клеток из уже синтезированной в цитоплазме клетки пальмитиновой кислоты или из жирных кислот экзогенного происхождения, т.е. поступающих из кишечника, образуются жирные кислоты, содержащие 18, 20 и 22 углеродных атома.
Иными словами, митохондриальная система биосинтеза жирных кислот, включающая несколько модифицированную последовательность реакций β-окисления, осуществляет только удлинение существующих в организме среднецепочечных жирных кислот, в то время как полный биосинтезпальмитиновой кислоты из ацетил-КоА активно протекает в цитозоле, т.е. вне митохондрий, по совершенно другому пути.
Внемитохондриальная система биосинтеза de novo жирных кислот (ли-погенез). Эта система находится в растворимой (цитозольной) фракции клеток многих органов, в частности печени, почек, мозга, легких, молочной железы, а также в жировой ткани. Биосинтез жирных кислотпротекает с участием НАДФН, АТФ, Мn2+ и НСО3– (в качестве источника СО2); субстратом является ацетил-КоА, конечным продуктом –пальмитиновая кислота. Потребности в кофакторах процессов биосинтеза и β-окисления жирных кислот значительно различаются.
Как отмечалось, строительным блоком для синтеза жирных кислот в цитозоле клетки служит ацетил-КоА, который в основном поступает измитохондрий. Было выявлено, что цитрат стимулирует синтез жирных кислот в цитозоле клетки. Известно также, что образующийся в митохондриях в процессе окислительного декарбоксилирования пирувата и окисления жирных кислот ацетил-КоА не может диффундировать в цито-золь клетки, так как митохондриальная мембрана непроницаема для данного субстрата. Поэтому вначале внутримитохондриальный ацетил-КоА взаимодействует с оксалоацетатом, в результате чего образуется цитрат. Реакция катализируется ферментом цитрат-синтазой. Образовавшийся цитрат переносится через мембрану митохондрий в цитозоль при помощи специальной трикарбоксилаттранспортирующей системы.
В цитозоле цитрат реагирует с HS-KoA и АТФ, вновь распадаясь на ацетил-КоА и оксалоацетат. Эта реакция катализируется АТФ-цитрат-лиа-зой. Уже в цитозоле оксалоацетат при участии цитозольной малатдегидро-геназы восстанавливается до малата. Последний при помощи дикарбокси-латтранспортирующей системы возвращается в митохондриальный мат-рикс, где окисляется до оксалоацетата, завершая тем самым так называемый челночный цикл:

Существует еще один путь переноса внутримитохондриального аце-тил-КоА в цитозоль клетки – с участием карнитина. Как отмечалось, кар-нитин играет роль переносчика ацильных групп из цитозоля в митохондрии при окислении жирных кислот. По-видимому, он может выполнять эту роль и в обратном процессе, т.е. в переносе ацильных радикалов, в том числе ацетильного радикала, из митохондрий в цитозоль клетки. Однако, когда речь идет о синтезе жирных кислот, данный путь переноса ацетил-КоА не является главным.
Образование малонил-КоА. Первой реакцией биосинтеза жирных кислот является карбоксилирование ацетил-КоА, для чего требуются бикарбонат, АТФ, ионы марганца. Катализирует эту реакцию фермент ацетил-КоА-кар-боксилаза. Фермент содержит в качестве простетической группы биотин. Авидин – ингибитор биотина угнетает эту реакцию, как и синтез жирных кислот в целом.
Установлено, что ацетил-КоА-карбоксилаза состоит из переменного числа одинаковых субъединиц, каждая из которых содержит биотин, биотинкарбоксилазу, карбоксибиотинпереносящий белок, транскарбоксилазу, а также регуляторный ал-лостерический центр, т.е. представляет собой полиферментный комплекс.
Реакция протекает в два этапа: I – карбоксилирование биотина с участием АТФ и II – перенос карбоксильной группы на ацетил-КоА, в результате чего образуется малонил-КоА:

Малонил-КоА представляет собой первый специфический продукт биосинтеза жирных кислот. В присутствии соответствующей ферментной системы малонил-КоА быстро превращается в жирные кислоты.
Энзиматические системы, осуществляющие синтез жирных кислот, называются жирно-кислотными синтетазами. Они широко встречаются в природе и могут быть изолированы из различных одноклеточных организмов, растений и животных тканей. Жирно-кислотные синтетазы делятся на 2 группы. К первой группе относятся полиэнзимные, не поддающиеся фракционированию комплексы с мол. м. порядка 500000, в которых все индивидуальные энзимы собраны в компактную структуру. В частности, в эту группу входят жирно-кислотныесинтетазы животных тканей и дрожжей. Вторая группа включает жирно-кислотные синтетазы, из которых отдельные энзимы могут быть выделены методами белкового фракционирования. Такие синтетазы встречаются у ряда микроорганизмов (в частности, у E.coli) и растений. Иными словами, в этих случаях все индивидуальныеферменты синтетазной системы находятся в виде автономных полипептидов. Мультиферментный комплекс, называемый синтетазой (синтазой) жирных кислот, состоит из 6 ферментов, связанных с так называемым ацилпереносящим белком (АПБ). Этот белок относительно термостабилен, имеет две свободные HS-группы (цистеина и фосфопантетеинового остатка, присоединенного к ОН-группе серина) и вовлекается в процесс синтеза высших жирных кислот практически на всех его этапах. Мол. масса АПБ составляет около 10000. Данный белок в синтетазной системе выполняет роль КоА. Заметим, что в животных тканях не удалось обнаружить свободного АПБ, подобного микробному. Из печени выделен полиэнзимный комплекс, содержащий все энзимы, необходимые для синтеза жирных кислот. Энзимы комплекса настолько прочно связаны друг с другом, что все попытки изолировать их в индивидуальном виде не увенчались успехом. Приводим последовательность реакций, происходящих при синтезе жирных кислот:

Далее цикл реакций повторяется. Допустим, что идет синтез пальмитиновой кислоты (С16). В этом случае образованием бутирил-АПБ завершается лишь первый из 7 циклов, в каждом из которых началом является присоединение молекулы малонил-АПБ к карбоксильному концу растущей цепижирной кислоты. При этом отщепляется дистальная карбоксильная группа малонил-АПБ в виде СО2. Например, образовавшийся в первом цикле бутирил-АПБ взаимодействует с малонил-АПБ:

Завершается синтез жирной кислоты отщеплением HS-АПБ от ацил-АПБ под влиянием фермента деацилазы. Например:

Суммарное уравнение синтеза пальмитиновой кислоты можно записать так:

Или, учитывая, что на образование одной молекулы малонил-КоА из ацетил-КоА расходуются одна молекула АТФ и одна молекула СО2, которая затем отщепляется, суммарное уравнение можно представить в следующем виде:

По сравнению с β-окислением биосинтез жирных кислот имеет ряд характерных особенностей: синтез жирных кислот в основном осуществляется в цитозоле клетки, а окисление – в митохондриях; участие в процессе биосинтеза жирных кислот малонил-КоА, который образуется путем связывания СО2 (в присутствии биотин-фермента и АТФ) с ацетил-КоА; на всех этапах синтеза жирных кислот принимает участие ацилпереносящий белок (HS-АПБ); при биосинтезе образуется D(–)-изомер 3-гидроксикис-лоты, а не L(+)-изомер, как это имеет место при β-окислении жирных кислот; необходимость для синтеза жирных кислот кофермента НАДФН. Последний в организме частично (на 50%) образуется в реакциях пен-тозофосфатного цикла, частично – в других реакциях, в частности в реакциях:
Малат + НАДФ+-> Пируват + С02 + НАДФН + Н+ Изоцитрат + НАДФ+-> α-Кетоглутарат + С02 + НАДФН + Н +.
3. Метаболизм АК с разветвленной цепью.
ВСАА (иначе branch chain amino acids) - это аминокислоты с разветвленными углеродными цепями. Точнее, это три аминокислоты: валин, лейцин и изолейцин. Они относятся к категории незаменимых, то есть организм не может их синтезировать самостоятельно. ВСАА метаболизируются в мышцах и действуют как переносчики азота при синтезе заменимых аминокислот, а также веществ, необходимых для протекания анаболических процессов. После того, как в ваш организм поступает пища, наиболее быстро усваивающимися аминокислотами являются ВСАА. Каждая из этих аминокислот вносит 10% вклада в обеспечение мышц энергией во время интенсивных упражнений; особенно данный эффект проявляется при низкокалорийной диете. Изолейцин участвует в образовании гликогена и гемоглобина, утилизации холестерина, в метаболизме Сахаров, валин участвует в образовании и запасании гликогена, в синтезе пантотеновой кислоты, изолейцин участвует в утилизации холестерина и метаболизме Сахаров. Валин - незаменимая аминокислота, оказывающая стимулирующее действие. Валин необходим для метаболизма в мышцах, восстановления поврежденных тканей и для поддержания нормального обмена азота в организме. Относится к разветвленным аминокислотам, и это означает, что он может быть использован мышцами в качестве источника энергии. Валин часто используют для коррекции выраженных дефицитов аминокислот, возникших в результате привыкания к лекарствам. Чрезмерно высокий уровень валина может привести к таким симптомам, как парестезии (ощущение мурашек на ко же), вплоть до галлюцинаций. Лейцин - незаменимая аминокислота, относящаяся к трем разветвленным аминокислотам. Действуя вместе, они защищают мышечные ткани и являются источниками энергии, а также способствуют восстановлению костей, кожи, мышц. Поэтому их прием часто рекомендуют в восстановительный период после травм и операций. Лейцин также несколько понижает уровень сахара в крови и стимулирует выделение гормона роста. Изолейцин — незаменимая аминокислота, относящаяся к трем разветвленным аминокислотам. Изолейцин необходим при многих психических заболеваниях; дефицит этой аминокислоты приводит к возникновению симптомов, сходных с гипогликемией. Необходим для синтеза гемоглобина. Также стабилизирует и регулирует уровень сахара в крови и процессы энергообеспечения. Метаболизм изолейцина происходит в мышечной ткани.
Аминокислоты с разветвленной цепью (АКРЦ) -валин, лейцин, изолейцин - при катаболизме превращаются в a–кетокислоты (оксикислоты с разветвленной цепью - ОКРЦ). -NH3
АКРЦ ® ОКРЦ
Этапы окисления АКРЦ:
1) трансаминирование:
АКРЦ + a–КГ ® ОКРЦ + Глу.
Фермент - АКРЦ–аминотрансфераза.
Наибольшая активность этого фермента наблюдается в сердце, почках, меньше – в скелетных мышцах, самая низкая – в печени;
2) дегидратация ОКРЦ до промежуточных продуктов ЦЛК. Фермент - дегидрогеназа ОКРЦ – локализован во внутренней мембране митохондрий и катализирует реакцию окислительного декарбоксилирования, в результате которой образуются промежуточные продукты ЦЛК:
Лей ® ацетил–КоА и ацетоацетат.
Вал, Иле ® сукцинил–КоА.
Катаболизм Вал и Иле (как и Мет) до сукцинил–КоА сопровождается образованием пропионил–КоА и метилмалонил–КоА:
|

 СО2 , вит. Н
СО2 , вит. Н
 Пропионил–КоА Метилмалонил–КоА
Пропионил–КоА Метилмалонил–КоА
 1
1
вит. В12
Сукцинил–КоА.
Ферменты, которые катализируют указанные реакции:
1) пропионил КоА–карбоксилаза (кофермент - биотин);
2) матилмалонил КоА–мутаза (кофермент -дезоксиаденозилкобаламин).
При дефиците В12 нарушается превращение метилмалонил-КоА в сукцинил-КоА, следствием которого является выведение большого количества метилмалоновой кислоты с мочей - метилмалоновая ацидурия. Метилмалоновая кислота токсична для нервной ткани и при отсутствии лечения вызывает дегенерацию заднебоковых столбов спинного мозга.
Билет №25
1. Репликация ДНК
Реплика́ция ДНК — процесс синтеза дочерней молекулы дезоксирибонуклеиновой кислоты на матрице родительской молекулы ДНК. Этот процесс обеспечивает точную передачу генетической информации из поколения в поколение. По имеющимся данным, в репликации ДНК, включающей узнавание точки начала процесса, расплетение родительских цепей ДНК в репликационной вилке, инициацию биосинтеза дочерних цепей и дальнейшую их элонгацию и, наконец, окончание (терминация) процесса, участвует более 40 ферментов и белковых факторов, объединенных в единую ДНК-репликазную систему, называемую реплисомой.
Цепи молекулы ДНК расходятся, образуют репликационную вилку, и каждая из них становится матрицей, на которой синтезируется новая комплементарная цепь. В результате образуются две новые двуспиральные молекулы ДНК, идентичные родительской молекуле.
Ферменты хеликазы (расплетают концы), топоизомеразы (раскручивают суперспирализованные витки) и ДНК-связывающие белки расплетают ДНК, удерживают матрицу в разведённом состоянии и вращают молекулу ДНК. Правильность репликации обеспечивается точным соответствием комплементарных пар оснований и активностью ДНК-полимераз, способных распознать и исправить ошибку. Источниками энергии и одновременно с этим субстратами являются dATP, dGTP, dTTP, dCTP. У прокариот выделено три типа ДНК-полимераз. Функцию элонгации выполняет ДНК-полимераза ІІІ. ДНК-полимеразы І и ІІ выполняют репарационные функции. Для затравки (инициации) требуется олигорибонуклеотид, который синтезируется праймазой.
Свойства процесса репликации:
1) матричный — последовательность синтезируемой цепи ДНК однозначно определяется последовательностью материнской цепи в соответствии с принципом комплементарности;
2) полуконсервативный — одна цепь молекулы ДНК, образовавшейся в результате репликации, является вновь синтезированной, а вторая — материнской;
3) идёт в направлении от 5’-конца новой молекулы к 3’-концу;
4) полунепрерывный — одна из цепей ДНК синтезируется непрерывно, а вторая — в виде набора отдельных коротких фрагментов (фрагментов Оказаки);
5) начинается с определённых участков ДНК, которые называются сайтами инициации репликации (англ. origin).

Сложность процесса репликации ДНК объясняется тем, что обе цепи реплицируются одновременно, хотя имеют разное направление (5'–>3' и 3'–>5'); кроме того, рост дочерних цепей также должен происходить в противоположных направлениях. Элонгация каждой дочерней цепи может осуществляться только в направлении 5'–>3'. Р. Оказаки высказал предположение, подтвержденное экспериментальными данными, что синтез одной из дочерних цепей осуществляется непрерывно в одном направлении, в то время как синтез другой дочерней цепи происходит прерывисто, путем соединения коротких фрагментов (в честь автора названы фрагментами Оказаки), в свою очередь синтезирующихся в противоположном направлении.
Как видно, синтез ведущей цепи ДНК идет всегда в направлении 5'–>3', соответствующем направлению движения репликационной вилки. Сохраняя правило синтеза дочерних молекул ДНК 5'–>3', синтез на второй цепи родительской ДНК идет в направлении, противоположном движению репликационной вилки. В зависимости от типа клетки фрагменты Оказаки имеют разные размеры – от нескольких сот до нескольких тысяч нуклеотидов (150–200 у эукариот и 1000–2000 у бактерий).
 Образование каждого фрагмента Оказаки требует наличия короткого затравочного комплементарного праймера – участка РНК, синтез которого катализируется праймазой. Затем при участии ДНК-полимеразы III синтезируются длинные участки ДНК. РНК-затравки далее вырезаются при участии ДНК-полимеразы I, а свободные места их (бреши) замещаются (достраиваются) комплементарными дезоксирибонуклеотидами под действием той же ДНК-полимеразы I; наконец, сшивание разъединенных участков отстающей цепи осуществляется при помощи ДНК-лигаз.
Образование каждого фрагмента Оказаки требует наличия короткого затравочного комплементарного праймера – участка РНК, синтез которого катализируется праймазой. Затем при участии ДНК-полимеразы III синтезируются длинные участки ДНК. РНК-затравки далее вырезаются при участии ДНК-полимеразы I, а свободные места их (бреши) замещаются (достраиваются) комплементарными дезоксирибонуклеотидами под действием той же ДНК-полимеразы I; наконец, сшивание разъединенных участков отстающей цепи осуществляется при помощи ДНК-лигаз.
По имеющимся данным, в репликации ДНК, включающей узнавание точки начала процесса, расплетение родительских цепей ДНК в репликационной вилке, инициацию биосинтеза дочерних цепей и дальнейшую их элонгацию и, наконец, окончание (терминация) процесса, участвует более 40 ферментов и белковых факторов, объединенных в единую ДНК-репликазную систему, называемую реплисомой. Рассмотрим подробнее её компоненты в прокариотической клетке:
Основным ферментом, катализирующим биосинтез новообразованной ДНК (стадию элонгации репликации ДНК), являются ДНК-полимеразы III. Имеются доказательства, что в димерной форме ДНК-полимераза III катализирует сопряженный синтез ведущей (лидирующей) и отстающей цепей ДНК при репликации.
ДНК-полимеразы I катализирует отщепление затравочного олигорибонуклеотидного праймера и заполнение образующихся после этого пробелов (ниш) дезоксирибонуклеотидами. ДНК-полимеразы II из Е. coli выполняет ≪ремонтные≫ функции, исправляя повреждения цепей ДНК.
Функцию раскручивания (расплетения) двойной спирали ДНК в репликационной вилке, происходящего за счет энергии гидролиза АТФ, выполняет специфический rep-белок, названный хеликазой. Образовавшиеся на определенное время одноцепочечные участки ДНК служат в качестве матрицы при репликации и стабилизируются при помощи особых белков, связывающихся с одноцепочечной ДНК (SSB-белки) и препятствующих обратному комплементарному взаимодействию цепей ДНК. В связи с этим их иногда называют дестабилизирующими двойную спираль белками. Имеются, кроме того, особые ферменты топоизомеразы (у прокариот одна из них названа ДНК-гиразой), которые играют особую роль в сверхспирализации, обеспечивая как репликацию, так и транскрипцию ДНК. Эти ферменты наделены способностью не только создавать супервитки, но и уничтожать суперспирализацию путем сшивания образующихся разрывов или разрезания ДНК.
В стадии инициации репликации ДНК участвует специфическая клеточная РНК-полимераза, названная праймазой, которая катализирует синтез короткого олигорибонуклеотида (от 10 до 60 нуклеотидов), т.е. праймера, с которого затем начинается синтез ДНК. В состав праймасомы входит также комплекс белков dna В и dna С, который вблизи репликационной вилки периодически участвует в формировании специфической вторичной структуры ДНК, подходящей для узнавания праймазой.
Важную функцию соединения двух цепей ДНК или замыкания двух концов одной цепи ДНК в процессе репликации либо репарации ДНК выполняют особые ферменты – ДНК-лигаза, катализирующая за счет энергии АТФ образование фосфодиэфирной связи между 3'-ОН-группой дезоксирибозы одной цепи и 5'-фосфатной группой другой цепи ДНК.

