




Симптомы синдрома Дауна
Симптомы синдрома Дауна можно увидеть уже при появлении на свет новорожденного ребенка. Ярко выражены и отчетливо видны во внешности характерные физические отличия: плоское лицо, косой разрез глаз, складка на верхнем веке, неправильная форма черепа, плоский затылок, маленькие ушные раковины, короткие нижние и верхние конечности, короткие пальцы, искривленный мизинец, имеется поперечная складка на ладони.
Слово «синдром» подразумевает наличие определенных признаков или характерных черт. Синдром Дауна впервые описал в 1866 году британский врач Джон Лэнгдон Даун. Почти сто лет спустя, в 1959 году, французский ученый Жером Лежен обосновал генетическое происхождение этого синдрома, которое определяется наличием в клетках человека дополнительной хромосомы.
Наличие этой генетической аномалии обуславливает появление ряда особенностей, вследствие которых ребенок будет медленнее развиваться и позже своих ровесников проходить общие для всех детей этапы развития. Таким малышам труднее учиться.
Как их называют «солнечные дети» часто имеют пороки сердца, для них характерны торможение и искажение роста костей, умственная отсталость, сниженный мышечный тонус, нарушение координации движений. Такие ребята позже начинают ходить, говорить, но у многих из них есть музыкальный слух, они очень веселые, ласковые и услужливые. И все же подавляющее большинство детей, тем более когда предприняты ранние и последовательные усилия по закреплению моторных навыков и общему развитию ребенка приводят к хорошим результатам: ребенок не только обслуживает себя самостоятельно, учиться ходить, есть, одеваться, говорить, играть и дружить со сверстниками, способен посещать специализированную или даже обычную школу,
заниматься спортом, словом делать то, что умеют делать другие дети,
Лечение
В медикаментозном лечении, а порой и оперативном вмешательстве, нуждается не синдром Дауна, а сопровождающие его заболевания. Чтобы избежать опасных осложнений, ребенок должен быть под постоянным контролем целого ряда узких специалистов.
Когда ребенок с синдромом Дауна здоров, он нуждается в развивающих, общеукрепляющих и обучающих программах, коррекционных занятиях со специалистами логопедами-дефектологами.
Выбор обучающих тренингов остается на усмотрение педиатров, педагогов и родителей ребенка с синдромом Дауна. Множество современных и уже опробованных методик дают прекрасные результаты.Например, анималотерапия отличается формированием особо доверительного контакта больного ребенка и специально подготовленного животного. Игровая форма обучения фиксирует интерес ребенка, снижает его утомляемость. В тесном контакте с животным развиваются моторные навыки, тренируются слабые мышцы, расширяется кругозор и улучшается психоэмоциональное состояние ребенка с синдромом Дауна.
В ряду особо перспективных обучающих программ с участием животных, стоит дельфинотерапия. Специально обученные дельфины погружают ребенка в мир чуткого тактильного контакта и ультразвукового общего воздействия. Игры и занятия с «улыбчивыми» дельфинами развивают мышечный корсет ребенка, его моторные и речевые навыки. Ультразвук из сонара дельфина благотворно воздействует на центральную нервную систему, укрепляет иммунитет, улучшает функцию внутренних органов и эндокринных желез.
Занятия с дельфинами обучают больного ребенка самостоятельности и независимости от посторонней помощи. Ребенок запоминает позитивные модели поведения в незнакомой обстановке и в дальнейшем легче адаптируется в социуме.
Продолжительность жизни при синдроме Дауна зависит от многих факторов и в благоприятных обстоятельствах человек проживает 50 лет и больше.
_____________
Также сюда относится синдром Прадера-Вилли
В организме здоровых людей присутствует копия генов, благодаря которой он может работать без отклонений от нормы. При синдроме Прадера-Вилли такая копия отсутствует. Первыми, кто исследовал и описал признаки данного заболевания в 1956 году, были Андреа Прадери Генрих Вилли. Также исследованиями занимались Алексис Лабхарт, Эндрю Зиглером и Гвидо Фанкони, которые также занимались изучением этого хромосомного нарушения и внесли свой вклад.
Синдром Прадера-Вилли (сокращенно СПВ) - это редкое генетическое заболевание, при котором семь генов (или некоторые их части) на 15 отцовской хромосоме (Q 11-13) - удалены или нормально не функционируют (например, при частичной делеции хромосомы 15Q). То есть наследственная информация, содержащаяся в ДНК, не преобразуется в РНК. При диагностике и профилактике стоит помнить о том, что причиной синдрома Прадера-Вилли может являться только экспрессия отцовских генов.
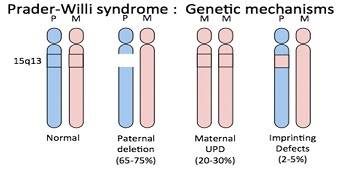
При этом заболевании имеет место дисфункция гипоталамуса, которая преимущественно отмечается в двух его ядрах – вентромедиального и вентролатерального.
Дисфункция гипоталамуса — симптомокомплекс, выражающийся в полигландулярной дисфункции с нарушением обменных и трофических процессов, менструального цикла и сопровождающийся нарушениями сердечнососудистой и нервной систем. Это приводит к сбоям в развитии вторичных половых признаков, то есть, к гипогонадизму, который развивается по гипогонадотропному типу. Пониженная активность тироназы в меланоцитах и фолликулах волос приводит к тому, что кожа, волосы и радужка глаза становятся гипопигментированными.
Для них также характерна пониженная концентрация соматолиберина. Это обусловлено тем, что 15-я хромосома связана с гипоталамусом. Однако при вскрытии умерших с синдромом Прадера — Вилли не было обнаружено никаких дефектов гипоталамуса. По другим данным, наблюдалось снижение общего числа клеток и окситоцин-содержащих клеток паравентрикулярных ядер гипоталамуса

Для синдрома Прадера — Вилли характерны:
- до рождения: низкая подвижность плода;
- часто — неправильное положение плода;
- дисплазия тазобедренных суставов
- ожирение; склонность к перееданию (чаще проявляется к 2-м годам);
- пониженный мышечный тонус (гипотонус); пониженная координация движений;
- маленькие кисти и стопы, низкий рост;
- повышенная сонливость;
- страбизм (косоглазие);
- сколиоз (искривление позвоночника);
- пониженная плотность костей;
- густая слюна; плохие зубы;
- сниженная функция половых желёз (гипогонадизм); в результате, как правило, бесплодие;
- речевая задержка, задержка психического развития; отставание в освоении навыков общей и мелкой моторики.
- более позднее половое созревание.
Внешние признаки: у взрослых выражена переносица; лоб высокий и узкий; глаза, как правило, миндалевидные; губы узкие.
Как правило, у больных встречается не более пяти вышеуказанных признаков.
Симптомы заболевания
 Данный синдром можно обнаружить еще на ранних сроках беременности. УЗИ диагностика позволит заметить неправильное расположение плода и его малую подвижность. К тому же у беременной существенно меняется уровень гормона гонадотропина, который вырабытывается клетками хориона. К внутриутробным признакам, помимо пониженной активности плода, также относят его ненормальное положение и многоводие.
Данный синдром можно обнаружить еще на ранних сроках беременности. УЗИ диагностика позволит заметить неправильное расположение плода и его малую подвижность. К тому же у беременной существенно меняется уровень гормона гонадотропина, который вырабытывается клетками хориона. К внутриутробным признакам, помимо пониженной активности плода, также относят его ненормальное положение и многоводие.
У младенцев синдром Прадера-Вилли выражается в наличии врожденного вывиха бедра (дисплазии), ослаблении тонуса мышц, а также в нарушении координации. Бывают случаи, когда ребенок не в состоянии самостоятельно сосать и глотать грудное молоко, так что питание осуществляется с помощью зонда. Также могут возникать проблемы с дыханием, которые могут быть настолько серьезными, что может потребоваться искусственная вентиляция лёгких.
Диагностика
Синдром диагностируется путём генетического анализа, рекомендуемого для новорождённых с пониженным мышечным тонусом (гипотонусом). Иногда вместо диагноза «синдром Прадера — Вилли» врачи ошибочно ставят диагноз «синдром Дауна» (поскольку синдром Дауна встречается намного чаще).
Дети с синдромом Прадера-Вилли очень похожи между собой, опытный генетик сможет быть уверен в диагнозе, не дожидаясь результатов исследования кариоти
Помимо всего вышеперечисленного, есть и прочие симптомы синдрома Прадера-Вилли. Так, например, у ребенка может наблюдаться повышенная сонливость. Что касается внешних признаков, то у него наблюдается задержка в развитии, поэтому для таких пациентов характерен низкий рост, а также маленькие кисти и стопы. Нередко бывает косоглазие. Для постановки диагноза существует ряд критериев, которые разделяют на большие и малые.
Перспективы развития
У большинства людей с синдромом Прадера — Вилли наблюдается задержка психического и речевого развития. Согласно исследованиям Керфс и Фрим (1992),
- 5 % обследованных продемонстрировали средний уровень коэффициента интеллекта (более 85 баллов по шкале IQ);
- 27 % — уровень на грани среднего (70—85 баллов);
- 34 % — уровень слабого отставания (50—70 баллов);
- 27 % — уровень среднего отставания (35—70 баллов);
- 5 % — сильное отставание (20—35 баллов);
- менее 1 % — значительное отставание.
По другим исследованиям (Кэссиди), 40 % пациентов с синдромом Прадера-Вилли демонстрируют интеллект на грани среднего или сниженный уровень интеллекта.
Как правило, дети с синдромом Прадера-Вилли имеют хорошую долговременную зрительную память, они могут научиться читать, могут обладать богатым пассивным словарём, но их собственная речь обычно хуже, чем понимание. Слуховая память, математические навыки и навыки письма, зрительная и слуховая кратковременная память у таких детей обычно значительно хуже.Синдром Прадера — Вилли нередко ассоциируется с повышенным аппетитом. У больных повышена концентрация в крови гормона грелина.
В настоящее время существует заболевание, которое по своей сути аналогично болезни Прадера-Вилли. Подобный механизм возникновения наблюдается при синдроме Ангельмана, только в этом случае мутация поражает материнский генетический материал. Эти заболевания, как правило, проявляются в различном виде и имеют разные формы и степень тяжести, но оба они неизлечимы.

синдром Альгельмана

Синдром Ангельмана – генетическая аномалия. Это довольно редкое заболевание, вызванное хромосомными нарушениями и встречающееся примерно у 1 из 10000 новорожденных. Для неё характерны задержка психического развития, нарушения сна, припадки, хаотические движения (особенно рук), частый смех или улыбки. При синдроме Ангельмана отсутствуют некоторые гены из длинного плеча 15-й хромосомы (в большинстве случаев — частичная делеция либо другая мутация 15 хромосомы). При синдроме Ангельмана страдает материнская хромосома; в случае повреждения отцовской хромосомы возникает синдром Прадера-Вилли.
Для синдрома Ангельмана характерны особенности:
- В 75 % проблемы с питанием, особенно с грудным вскармливанием, такие младенцы плохо набирают вес;
- задержка в развитии навыков общей моторики (умение сидеть, ходить);
- задержка речевого развития, неразвитая речь (у всех детей);
- дети больше понимают, чем могут сказать или выразить;
- дефицит внимания и гиперактивность;
- сложности с обучением;
- эпилепсия (80 % случаев), нарушения выявляются также при электроэнцефалографии; считается, что у детей с синдромом Ангельмана наблюдается вторичная (симптоматическая) эпилепсия;
- необычные движения (мелкий тремор, хаотические движения конечностей);
- частый смех без повода;
- ходьба на негнущихся ногах — из-за этой особенности детей с этим синдромом иногда сравнивали с марионетками;
- размер головы меньше среднего, нередко с уплощением затылка;
- иногда характерные черты лица — широкий рот, редко расположенные зубы, выдающийся вперед подбородок, высунутый наружу язык;
- нарушения сна;
- страбизм (косоглазие) в 40 % случаев;
- сколиоз (искривление позвоночника) в 10 % случаев;
- повышенная чувствительность к высокой температуре;
- чувствуют себя комфортнее в воде.
Диагностика
Синдром диагностируется путём генетического анализа (15 хромосома), рекомендуемого для новорожденных с пониженным мышечным тонусом (гипотонусом), отставанием в развитии общей моторики и в развитии речи. Родители и врачи должны обратить внимание на случаи мелкого тремора, хаотические, порывистые движения конечностей, походку с негнущимися ногами; в ряде случаев специфическое выражение лица, слишком частый смех.
Так как речь идет о генетическом дефекте, медикаментозного лечения для него не существует, однако есть процедуры, которые способны улучшить качество жизни пациентов с диагнозом «синдром Ангельмана». Например, специалисты с раннего детства рекомендуют делать больному массаж и физпроцедуры для укрепления организма.
Перспективы развития
Дети с синдромом Ангельмана понимают намного больше, чем могут сказать. В некоторых случаях у них вообще нет речи; описаны дети со словарным запасом около 5-10 слов. При этом люди с синдромом Ангельмана любят общаться с другими людьми, играть, как правило, они дружелюбны.
Рекомендуется обучать таких детей языку жестов. Занятия с раннего возраста по специальным программам, направленные на развитие навыков мелкой и общей моторики, в ряде случаев дают хорошие результаты.

Перспективы развития зависят от степени поражённости хромосомы. Некоторые люди с синдромом Ангельмана способны освоить навыки самообслуживания и речь на примитивном уровне (обычно причиной синдрома в этом случае стала мутация), некоторые никогда не смогут ходить и говорить (это обычно происходит в случае делеции части хромосомы).
С возрастом, как правило, симптомы гиперактивности и нарушения сна смягчаются. У девочек с синдромом Ангельмана в период полового созревания могут участиться припадки. Большинство людей с синдромом Ангельмана способны контролировать экскреторные функции (мочеиспускание и дефекацию) днем, некоторые — и ночью. Некоторые люди с синдромом Ангельмана способны есть при помощи ножа и вилки, одеваться самостоятельно в случае отсутствия на одежде пуговиц, «молний». Во взрослом возрасте может появиться ожирение и ухудшиться ситуация со сколиозом.
Менструации, половое созревание индивидов с синдромом Ангельмана происходит в обычные сроки. Описан один случай беременности женщины с синдромом Ангельмана: она родила девочку с таким же диагнозом.
При правильном уходе люди с данным расстройством могут дожить до взрослых лет, сами обслуживать себя, хотя они и не смогут никогда стать полностью самостоятельными.
_____________
А также синдром Вильямса.
Существует особое генетическое заболевание, при котором у человека возникают нарушения в работе мозга и других органов. Его называют двумя терминами: синдром Вильямса, "лицо эльфа".
Причины отклонения
Редкое генетическое нарушение, вызванное делецией участка, на котором находится около 26 генов и который расположен на длинном плече 7-й хромосомы (7q11.23)[2]., где находится около пятнадцати генов. Три гена, которые отсутствуют при данном заболевании, отвечают за нормальное развитие человеческого мозга и выработку эластина. Из-за последнего велика вероятность развития болезней сердца и сердечных сосудов. Такая патология получила название "синдром Вильямса". По исследованиям ученых, это заболевание встречается один раз на десять тысяч новорожденных.
Впервые на подобное заболевание обратил внимание врач-кардиолог Дж. Вильямс в начале шестидесятых годов прошлого столетия. Он обнаружил, что у ряда его пациентов, объединенных одинаковыми проблемами с сердцем и сосудами, есть и другие общие признаки: как во внешности, так и в умственном развитии. Проведя исследования, ученый установил, что причиной заболевания этого типа являются нарушения на генетическом уровне – в структуре хромосом.
Больные имеют особое строение лица, в специальной литературе называемое «лицом эльфа», поскольку оно напоминает лицо эльфов в их традиционном, фольклорном варианте. Для них характерны широкий лоб, разлёт бровей по средней линии, опущенные вниз полные щёки, большой рот с полными губами (особенно нижней), плоское переносье, своеобразная форма носа с плоским тупым концом, маленький, несколько заострённый подбородок.

Глаза зачастую ярко-голубые, со звёздчатой картиной радужки и склерами синеватого цвета. Разрез глаз своеобразный, с припухлостями вокруг век. Сходящееся косоглазие.
Для старших детей характерны длинные, редкие зубы. Обычно зубы у детей, страдающих этим заболеванием, прорезаются гораздо позже, а такие болезни, как кариес, встречаются чаще. Стоматолог, осматривающий таких детей, может обнаружить их недоразвитость, проблемы с корнями и прикусом.
С возрастом лицо больных несколько меняется: появляется массивность надбровных дуг, меньше выражена пастозность лица, нет плоского переносья и эпиканта. Обращает на себя внимание увеличенное расстояние от основания носа до верхней губы.
Сходство лиц усиливает улыбка, которая ещё больше подчёркивает отёчность век и своеобразное строение рта.
Встречается это заболевание как у мужчин, так и у женщин.
У больных синдромом Вильямса шея длинная и тонкая, грудная клетка уже, чем обычно, талия расположена ниже, ноги по форме напоминают букву Х. Зачастую у таких людей наблюдаются проблемы со стопами: неправильное их положение, вследствие чего развивается плоскостопие или косолапость. С возрастом, у детей с болезнью «эльфа» наблюдаются изменения во внешности, сильно грубеет и отекает лицо. У ребенка могут быть проблемы с координацией и работой опорно-двигательной системы. Если в раннем возрасте такие дети страдают излишней худобой и ослабленностью по сравнению со сверстниками, то со временем, напротив, у них возможно ожирение. Это связано со слабой сердечно-сосудистой системой.
Ни одна из этих черт не является обязательной, но их общее сочетание всегда присутствует.
Психологические особенности
Умственная отсталость при синдроме Вильямса наблюдается во всех случаях[1]. Для этого синдрома характерен дефицит наглядно-образного мышления, абстрактное мышление практически полностью отсутствует. Степень интеллектуального дефекта довольно значительна, IQ в среднем колеблется от 40 до 50 (умеренная умственная отсталость — имбецильность)[1].
Можно отметить большое сходство психопатологической картины дефекта у всех больных: при значительном снижении интеллекта больные имеют относительно большой словарный запас, очень словоохотливы, склонны к подражанию, речь у детей довольно хорошая, тем не менее она представляет всего лишь набор штампов. Вместе с тем всегда страдают пространственные представления, организация и планирование деятельности. Очень характерны и постоянны особенности личности этих детей: добродушие, приветливость, послушание, эмоциональность, стремление к общению, доверчивость[1]. Практически всегда имеется хороший музыкальный слух даже при выраженном интеллектуальном дефекте. Нередко выявляются неврозоподобные нарушения — энурез, привычная рвота, навязчивые действия, головная боль[1]. У части детей наблюдается гиперактивность, страхи и тревога[1].
Возможна частичная социальная адаптация. Некоторые дети могут учиться во вспомогательной школе, они овладевают чтением и письмом, но им недоступны действия, связанные с организацией даже простейших трудовых операций.

Помимо нарушений физического состояния организма, у детей, страдающих синдромом Вильямса, возникают проблемы и с мозговой деятельностью. У них низкая работоспособность, они не способны долго сосредотачиваться на поставленной задаче, плохо запоминают новый материал. Им трудно дается логика и пространственное мышление, в эмоциях и поступках они зачастую неадекватно себя ведут. У некоторых детей наблюдается истеричность, излишне инфантильное поведение, агрессия. В четверти случаев при эмоциональных вспышках возможны судороги.
Но не все дети, у которых обнаруживается болезнь "эльфа", ведут себя агрессивно. Зачастую такие дети очень общительные, доброжелательные, они веселые и доверчивые. Но в своих проявлениях эти люди слишком непосредственны и порой ведут себя неадекватно. В их поведении, даже во взрослом возрасте, можно заметить излишнюю детскость. Такие люди не самостоятельны, их реакции замедленны и в то же время хаотичны. Они нуждаются в поддержке со стороны других людей, их нужно подбадривать и воспитывать, чтобы помочь нормально адаптироваться в обществе.

Основное место занимают симптоматическое лечение и коррекционно-воспитательная работа, поэтому нужно попытаться максимально приспособить ребенка к окружающей жизни с помощью специальных программ.
Но даже в этом случае возможны трудности с обучением, поскольку таким детям трудно сосредоточиться, у них часто бывают проблемы с речью. Также у детей с синдромом Вильямса часто страдают внутренние органы: помимо сердечно-сосудистой, бывают нарушения мочеполовой системы, эндокринной. Зрение у людей с «лицом эльфа» во многих случаях ослабленное. У них нередки случаи таких заболеваний глаз, как колобома, атрофия зрительного нерва, катаракта.
На заметку:
Чтобы распознать у ребенка синдром Вильямса ("лицо эльфа"), родители должны обратить внимание не только на внешний вид, но и на особенности их развития в самом раннем возрасте. У таких детей организм более ослабленный, чем у сверстников, они отстают в развитии, как физически, так и интеллектуально. Аппетит у них плохой, их трудно заставить что-то съесть. При этом часто случаются расстройства желудка: рвота, диарея или запоры, желудочные колики. Если сделать анализ крови, то можно обнаружить, что уровень кальция и холестерина у больных повышен.
К трем годам физическое развитие детей с синдромом Вильямса приходит в норму, но можно заметить отклонения в речи. При этом ребенок живой, активный и общительный, потому родители не всегда могут понять, что у него есть проблемы. Также стоит обратить внимание на тембр голоса малыша. Если он слишком низкий, с явной хрипловатостью, это является одним из характерных признаков синдрома Вильямса.
Говорить такие дети начинают довольно поздно. По исследованиям ученых, впервые они произносят слова только к двум – трем годам, а составлять слова в фразы учатся лишь к четырем – пяти. У детей с болезнью «эльфа» нарушена моторика и зрительная координация. Движения неловкие и порывистые. Детям трудно освоить простейшие навыки, в том числе и самостоятельный уход за собой. В то же время у них наблюдается интерес к музыке, они с удовольствием ее слушают.
Крайне важно диагностировать синдром Вильямса уже на ранних стадиях, чтобы помочь детям адаптироваться к окружающей среде. Существуют специальные методики, которые могут помочь сделать это, учитывая особенности таких детей.
Подитог:
Причины умственной отсталости могут быть спровоцированы дисфункцией отдельных генов, а также числом мутаций генов в которых степень превышает 1000.
_____________________________________
Характеристика Олифрении
Заболевание относят к обширной группе болезней, связанных с нарушением в развитии. Олигофрениею считают аномалией недоразвития психики, личности, а также всего организма больного. Показатель олигофрении в индустриально развитых странах достигает 1% от всего населения, и из этого 85 % с легкой умственной отсталостью.
Соотношение заболевших мужчин к женщинам составляет 2:1.
Классификация Олигофрении
Существует несколько классификаций олигофрении.
Традиционно заболевание классифицируется по степени выраженности и
подразделяется на 3 вида:
- Дебильность (слабо выраженная)
- Имбецильность (слабо выраженная)


И Идиотия (сильно выраженная)

Однако, также существует альтернативная классификация по МКБ-10, по которой существует 4 степени тяжести:
Легкая
Умеренная
Тяжелая
Глубокая
| Рубрика (МКБ-10) | Степень умственной отсталости (МКБ-10) | Традиционный термин (МКБ-9) | Коэффициент интеллекта | Психологический возраст |
| F70 | Лёгкая | Дебильность | 50—69 | 9—12 лет |
| F71 | Умеренная | Нерезко выраженная имбецильность | 35—49 | 6—9 лет |
| F72 | Тяжёлая | Выраженная имбецильность | 20—34 | 3—6 лет |
| F73 | Глубокая | Идиотия | до 20 | до 3 лет |
Когда оценка степени умственной отсталости затруднена или невозможна (например, из-за глухонемоты, слепоты), используется категория F78«другие формы умственной отсталости».

