




Электрохимическая коррозия — это растворение металла в электролитах с появлением электрического тока (электронного в металле и ионного в электро-лите).
Как известно, электролитами могут быть различные кислоты,их водные растворы, щелочи и их растворы, растворы солей в воде (в том числе морская вода), а также вода, содержащая растворенныйк воздух.
Характерной особенностью электрохимической коррозии является то, что общая реакция химического взаимодействия электролита с металлом, напри-мер, цинка в растворе серной кислоты (Zn + H2SO4 = ZnSO4 + Н2), при электро-химической коррозии разделяется на два параллельных процесса: анодный - процесс перехода металла в раствор в виде гидротированных ионов с сохранением эквивалентного количества электронов в металле - и катодный – про-цесс ассимиляции этих электронов какими-либо деполяризаторами, т. е. ато-мами, молекулами или ионами раствора, которые могут восстанавливаться на катоде. Иначе говоря, один процесс - окислительный -растворение металла с отдачей электронов, а второй - восстановительный - выделение водорода или металла из раствора с присоединением электронов.
Большинство металлов и сплавов по природе своей неустойчивы и склон-ны к переходу в окисленное состояние. При контакте металла с агрессивной средой его ионы с кристаллической решетки переходят в реагент. Это энергети-чески выгодно, так как при этом понижается свободная энергия системы.
Интенсивность электрохимической коррозии металлов зависит от харак-тера раствора электролита (кислота, щелочь, раствор газов), концентрации это-го раствора и его температуры, а также от электрохимических свойств металла: от энергии выхода иона металла из кристаллической решетки в раствор, т. е. от потенциала растворения данного металла, или от так называемого нормального (стандартного) электродного потенциала.
Электрохимический потенциал представляет собой величину, пропорцио-нальную энергии перехода иона металла в электролит при данных условиях. Если два соединенных вместе различных металла опустить в электролит, то об-разуется гальваническая макропара с определенной разностью электрохимичес-ких (электродных) потенциалов.
Коррозия возможна в том случае, если металл, погруженный в электро-лит, обладает макро- или микронеоднородностью по химическому составу и физико-химическим свойствам, вследствие чего одни его места в данном элек-тролите будут обладать меньшим, а другие большим электрохимическим по-тенциалом, т. е. если металл в данных условиях будет представлять либо макро-скопический гальванический элемент, либо системы многоэлектродных микроскопических элементов.
Принципиальная схема электрохимического коррозионного разрушения металла приведена на рис. 22. Участки поверхности металла с более низким по-тенциалом оказываются анодами и растворяются, электроны при этом переме-щаются к участкам с более высоким (менее отрицательным или более положи-тельным, чем у данного анода) потенциалом (к катоду) и разряжаются на нем. Оба процесса (катодный и анодный) взаимно связаны и не могут протекать са-мостоятельно.
Причиной различия потенциалов стали могут быть включения, неравно-мерно распределенные примеси и легирующие элементы в металле, неравно-мерная газонасыщенность, неодинаковый наклеп металла, наличие в нем неско-льких различных по составу структурных составляющих, например, наличие карбидов или боридов в ферритной или аустенитной стали, феррита и аустени-та в двухфазной стали в случае воздействия на металл неокисляющей агрессив-ной среды и т. д. Причиной коррозии может быть также различие в электрохимических свойствах тела и границ зерен. При полностью однородном металле коррозия его может быть вызвана неоднородностью концентрации раствора, ностью напряжений в металле и т. д.
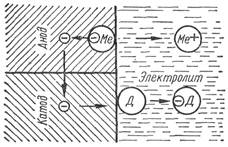
Рисунок 22 -Принципиальная схема электрохимического коррозионного процесса (по Томашову) (Д— катодный деполяризатор).
Однако в работах Н. Л. Томашова показано, что в действительности воз-никновение микроэлементов (гальванических пар) является только одним из возможных путей перехода системы в более термодинамически выгодное сос-тояние.
Для описания процесса коррозии и его кинетики следует пользоваться по-ляризационными кривыми, отображающими роль ведущих факторов процесса коррозии (рис. 23).
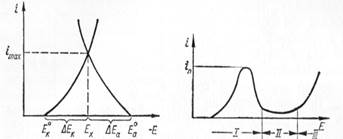
Рис. 23. Поляризационная диаграмма коррозии.
Рис. 24. Кривая анодной поляризации хрома в 1н. растворе H2SO4.
На рис. 23 по оси ординат отложен коррозионный ток (величина, пропор-циональная скорости коррозии), а по оси абсцисс - значение потенциалов элект-родов - Е. Если система разомкнута, то начальное значение потенциалов анода и катода будет равно соответственно Е°а и Ек0.Если катод и анод контакти-руют между собой (короткозамкнутая система), то их потенциалы сблизятся до общего значения EX а коррозионный ток системы будет соответствовать свое-му максимальному значению іmax. Таким образом, по величине коррозионного тока можно количественно судить об интенсивности коррозии. С другой сто-роны, поляризационные кривые позволяют судить о степени торможения про-текания катодного и анодного процессов. Соотношение между отрезками паде-ния потенциала вследствие анодной и катодной поляризации (∆Еа и ∆Ек) харак-теризует соотношение между анодным и катодным торможением.
Пассивность металлов — состояние относительно высокой коррозион-ной стойкости, вызванное торможением анодного процесса электрохимической коррозии в определенной области потенциалов. Переход металла из активного состояния в пассивное носит название пассивации.
Наступление пассивного состояния металла можно проследить по анод-ной поляризационной кривой, характер которой не зависит от истинной причи-ны пассивного состояния. На рис. 24 приведена анодная поляризационная кри вая, изображающая анодный процесс для хрома в 1 н. растворе H2SO4. Участок 1 кривой характеризует активное анодное растворение. Когда анодный ток дос-тигает своего предельного значения іnр начинается участок пассивации металла. Участок // кривой отвечает этому пассивному состоянию метала.
Несмотря на наличие защитного слоя, металл и в пассивированном сос-тоянии подвергается коррозионному разрушению, хотя и очень замедленному (например, у нержавеющих сталей коррозия составляет сотые доли миллиметра в год).
Существует две теории, объясняющие причину замедления анодного про-цесса - пленочная и адсорбционная. Обе теории исходят из представления об образовании «барьера» между металлом и коррозионной средой. Однако приро-да образования этого «барьера» (защитного слоя), образующего на металле во время перехода в пассивное состояние, до сих пор не выяснена.
Согласно пленочной теории роль «барьера» между металлом и коррози-онной средой выполняет тончайшая окисная пленка. Состав и строение этой пленки, образующейся в окислительных средах, а следовательно, и ее защитные свойства зависят от состава сплава. Так, на поверхности хромоникелевых ста-лей, кроме окислов железа, присутствует окисел хрома, а в хромоникельмолиб-деновой - и окисел молибдена.
Согласно другой теории, каталитическое защитное действие оказывает адсорбированный поверхностным слоем металла кислород].
Некоторые исследователи высказывают мнение, что наиболее правиль-ным механизмом пассивации следует считать пленочно-адсорбционный. При этом адсорбционный механизм торможения анодного процесса рассматривает-ся как вспомогательный.
При достижении определенного значения потенциала электрода (для хро-ма +1,3 ÷1,35 В ) начинается второй подъем кривой силы тока. Становится воз-можным переход металла в раствор с образованием высоковалентных ионов (участок ///); начинается область перепассивации.
Перепассивация металлов — нарушение пассивного состояния металла, сопровождающееся резким увеличением скорости его коррозии. При этом наб-людается сдвиг стационарного потенциала металла в сторону более положите-льных значений. Состояние перепассивации соответствует достижению потен-циала +1,3÷+1,35 В. Явление перепассивации объясняют] тем, что при опре-деленных условиях на поверхности металла образуются неустойчивые, легко растворяющиеся в реагенте соединения (окислы) высших валентностей. Сме-щение потенциала металла в сторону положительных значений может проис-ходить либо при повышении концентрации раствора и его температуры, либо при добавке к нему окислителя (например, К2Сг207 к раствору HNO3), либо при анодной поляризации (рис. 24).
Следует отметить, что для нержавеющих сталей при анодной поляриза-ции как в неокислительных средах (например, в растворах H2SO4), так и в окис-лительных растворах (HNO3 и HNO3 + + K2Cr207) при достижении области по-ложительных потенциалов + 1,3 ÷ +1,35 Внаблюдается увеличение коррози-онного тока, соответствующее процессу анодного растворения стали с пере-ходом в раствор ионов хрома высшей валентности. Таким образом, для начала состояния перепассивации любой нержавеющей стали, а также чистого хрома необходимо достижение одного и того же потенциала порядка + 1,3÷+1,35В
Виды коррозии
Коррозия может быть равномерна я и сосредоточенная. С целью обеспе-чения высокой работоспособности изделий в условиях воздействия агрессив-ных сред стремятся, чтобы металл не был склонен к сосредоточенной коррозии, а скорость равномерной коррозии по всей поверхности не превышала бы 0,1 г/м2 • чили глубина ее была бы не более 0,1 мм за год.
Различают несколько видов сосредоточенной коррозии: межкристал-литная - коррозия по пограничным слоям зерен; структурная – преимущест-венное растворение одной из фаз гетерофазного сплава; точечная - коррозия преимущественно в локализованных участках (точках) поверхности металла с распространением ее вглубь последнего.
Межкристаллитная коррозия - наиболее опасный вид коррозионного разрушения. Развиваясь по границам зерен, она распро- страняется в толщу металла (рис. 25).

Рис. 25. Межкристаллитная коррозия стали Х18Н10 после провоцирующего нагрева при температуре 6500 С в течение 2,5 ч, х150.
Металл, пораженный межкристаллитнои коррозией, разрушается от приложения даже незначительных нагрузок (рис. 26).
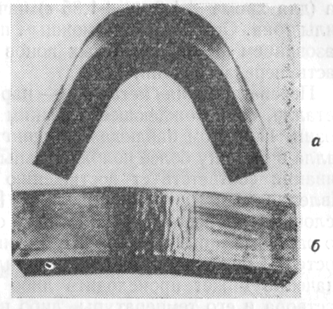
Рис. 26. Внешний вид изогнутых образцов стали Х18Н10, испытанных на склонность к межкристаллитнои коррозии по методу AM: а — в состоянии поставки (после аустенизации); б — после провоцирующего нагрева при 650° С в течение 2,5 ч.
Межкристаллитной коррозии подвержены, как правило, металлы, имею-щие однофазную структуру. Природа этого вида коррозии в настоящее время изучена недостаточно и существующие теории не во всех случаях позволяют объяснить причины, вызывающие восприимчивость металлов различного сос-тава к этому виду коррозии.
Стали, не восприимчивые к межкристаллитнои коррозии в состоянии поставки, могут приобрести склонность к ней после неблагоприятного терми-ческого воздействия, в том числе воздействия термического цикла сварки. При этом для аустенитных сталей опасным является нагрев в интервале 400-800° С, а для ферритных - нагрев выше 900° С с последующим быстрым охлаждением.
Существуют различные теории, объясняющие причину межкристаллит-ного коррозионного разрушения металла. Одна из наиболее распространенных коррозии аустенитных сталей - теория обеднения. Как известно, углерод обла-дает ограниченной растворимостью в аустените (0,02—0,03%, а по данным некоторых исследователей-даже до 0,007%).
В том случае, если содержание углерода превышает предел растворимос-ти, гомогенная аустенитная структура, образующаяся после высоко темпера-турной обработки (нагрев при температуре 1050-1150° С, последующее быст-рое охлаждение), находится в состоянии неустойчивого равновесия. При пов-торных нагревах в интервале температур 400-800° С она стремится к уменьше-нию свободной энергии, что приводит к выделению сложных карбидов хрома и железа [(Cr, Fe)3C или (Fe, Сг)23 Сб] по границам зерен (рис. 27).

Рис. 27. Микроструктура стали 08Х17Н5Г9АБ (Х600):
а - всостоянии поставки; б - после провоцирующего нагрева при 650° С в течение 2,5 ч.
Вместе с карбидами, по-видимому, выделяются и нитриды (Fe, Сг) N. Скорость выделений карбонитридов возрастает с повышением температуры и увеличением времени выдержки. При этом увеличивается и склонность к меж-кристаллитной коррозии. Выделяющиеся карбо-нитриды содержат значитель-ное количество хрома. При этом диффузия углерода из центральных участков зерен к границам протекает значительно быстрее, чем хрома, благодаря боль-шой разности коэффициентов диффузии их атомов (табл. 4).

Вновь поступивший в периферийные участки углерод образует карбиды, черпая запасы хрома в этих участках. Содержание хрома в них резко снижает-ся, достигая менее 12%. В результате этого сильно обедненные хромом пери-ферийные участки зерен теряют способность к пассивации и подвергаются ин-тенсивному коррозионному разрушению.
Рис. 28 иллюстрирует распределение хрома по зерну хромоникелевой аустенитной стали в состоянии поставки (после аустенитизации) и после провоцирующего нагрева.
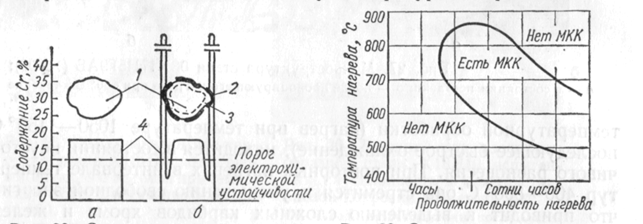
Рис. 28. Схема распределения хрома по зерну аустенитной стали типа 18-8, склонной к межкристаллитной коррозии:
а - в состоянии поставки (после аустенитизации); б - после нагрева в крити-ческом интервале температур:
1 - тело аустенитного зерна; 2 - карбиды хрома и железа по границам зерен;
3 - обедненные хромом пограничные слои аус-тенитных зерен; 4 - график распределения хрома.
Рис. 29. Схема влияния температуры и продолжительности нагрева на склонность нестабилизированных аустенитной стали и аустенитных швов к межкристаллитной коррозии.
При достаточно длительном нагреве металла в области критических тем-ператур стойкость его против межкрчсталлитной коррозии восстанавливается (рис. 29). При этом чем выше температура нагре-ва в интервале 400-800° С, тем быстрее восстанавливается стойкость против межкристаллитной коррозии. Кривая, ограничивающая область появления у стали склонности к межкристал-литной коррозии и отображающая зависимость между температурой и продолжительностью нагрева, носит название диаграммы Роллансона.
Восстановление стойкости металла против межкристаллитной коррозии обусловлено тем, что при длительной выдержке в области критических тем-ператур после выделения карбидов происходит выравнивание содержания хрома по телу зерна за счет его диффузии из центральных областей в пери-ферийные. Содержание хрома в обедненных участках повышается и они вновь приобретают способность к пассивации.
Некоторые авторы считают, что межкристаллитная коррозия может иметь место и при отсутствии обеднения границ зерен хромом. Любое выде-ление второй фазы, в том числе и карбидов, приводит к возникновению мик-ронапряжений, что в свою очередь влечет за собой более низкую поляризацию анодного процесса в пограничных областях между зернами и несовершенную их пассивацию.
Другие авторы высказывают мнение, что причиной межкристаллитной коррозии являются карбиды железа, выделяющиеся по границам зерен, которые отличаются очень низкой химической стойкостью.
Приведенные теории связывают появление склонности аустенитных ста-лей к межкристаллитной коррозии, наблюдаемой в восстановительной или слабоокислительной среде, главным образом с выделением второй фазы.
Однако сталь может быть склонна к межкристаллитному коррозионному разрушению и в том случае, если предварительно она прошла гомогенизирую-щую термическую обработку. Так, практически все хромоникелевые аустенит-ные стали склонны к межкристаллитной коррозии в сильноокислительных средах (например, в HNO3 концентрации выше 94%, в 65%-ной HNO3 с добавками К2Сг207 и т. д.), т. е. в том случае, если металл находится в области перепасси-вации.
Такое поведение металла, по-видимому, обусловлено изменением харак-тера образования защитного слоя на поверхности металла при переходе его из пассивного состояния в транспассивное. При испытании хромоникелевой стали и сварных швов типа 20-20 с различным содержанием кремния в кипящем 65%-ном растворе HNO3-c добавками К2Сг207 максимальная коррозия металла наблюдается у металла с 0,8—1,3% кремния (рис. 30)
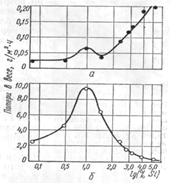
Рис. 30. Влияние кремния на общую коррозионную стойкость хро-
моникелевого наплавленного металла типа 20-20 в кипящих растворах
кислот: а - 32%-ная HNO8; б — 65%-ная HNO3 с добавкой 1 г/л К2Сг2О7.
Ферритные стали приобретают склонность к межкристаллитной коррозии после нагрева при Т > 900° С и последующего быстрого охлаждения. Как и для аустенитных сталей, в этом случае существуют различные трактовки причин, вызывающих эту склонность.
Некоторые авторы] считают, что при высоких температурах по границам ферритных зерен образуются аустенитные прослойки обедненные хромом. В процессе быстрого охлаждения они превращаются в мартенсит. Находясь при высоких температурах в равновесии с ферритом, пограничная фаза обогащается углеродом и обедняется хромом до уровня, при котором нарушается их пассив-ное состояние.
Очевидно, эта гипотеза справедлива для сталей, в которых наряду с фер-ритом имеется небольшое количество аустенитной фазы расположенной в виде прослоек между зернами.
Другие исследователи объясняют межкристаллитную коррозию феррит-ных сталей выпадением карбидов и нитридов непосредственно из пересыщен-ного углеродом и азотом феррита при охлаждении. Возникшее при этом нап-ряженное состояние в пограничных слоях зерен обусловливает потерю их стой-кости против коррозии
Существует мнение, что причиной межкристаллитной коррозии феррит-ных сталей после высокотемпературного нагрева и быстрого охлаждения явля-ется образование по границам зерен не стойких против воздействия агрессив-ных сред карбидов железа. Отпуск при температуре 700-800° С вызывает превращение их в карбиды хрома, обладающие стойкостью против коррозии. Некоторые исследователи [8, 141 считают эту гипотезу ошибочной. Содержание железа в карбидной фазе ферритных сталей в состоянии поставки после высокотемпературного нагрева с последующим быстрым охлаждением и после отпуска при различных температурах практически не изменяется (табл. 6).
Fe. 
Примечание. В состоянии поставки в стали 12X17 в карбидах железа и хрома содержится 0,508% Сг и 0.249% Fe, а в стали 15Х28АН - 2,77% Сг и 0,52% Fe.
Сторонники теории обеднения считают, что в процессе быстрого ох-лаждения ферритных сталей происходит выпадение вторичных фаз по гра-ницам зерен, обеднение пограничных областей хромом и, как следствие этого, снижение пассивности этих областей зерен. Указывается также, что, кроме обеднения границ зерен, определенную роль могут играть неравновесные карбиды с повышенным содержанием железа, а также напряжения, появившиеся в результате их выделения.
Есть основания полагать, что степень обеднения и его эффективность в электрохимическом растворении ферритного металла значительно меньше, чем аустенитного, так как скорость диффузии хрома, а следовательно, и выравни-вание его концентрации по телу зерна в первом выше, чем во втором.
По-видимому, при высокотемпературном нагреве хромистых ферритных сталей углерод и азот, находящиеся в структурно-свободных карбидах (карбо-нитридах), растворяются в феррите (преимущественно в пограничных слоях зерен) в количествах, значительно превышающих предел растворимости при комнатнойтемпературе. При последующем быстром охлаждении они частично, фиксируются в виде пересыщенного твердого раствора, вызывая перенапря-женность и искажение решетки. Неполное выделение карбидов (карбонитри-дов) при охлаждении металла, когда сохраняется когерентная связь их атомов с решеткой материнского твердого раствора, также вызывает дополнительные напряжения. При контактировании с реагентом напряженные и ненапряженные участки зерна образуют микроэлементы, в которых анодами служат напряжен-ные участки и соответственно легко корродируют. В ряде случаев при этом происходит локальная (сосредоточенная) коррозия как ферритного, так и аус-тенитного металла.
Действительно, выделение избыточной фазы, в том числе и карбидов, не-льзя представить иначе, как процесс, протекающий во времени и включающий несколько стадий. Первая стадия - накопление растворенных атомов (в данном случае углерода, азота, хрома, марганца) в пограничных зонах зерен до уровня состава новой фазы с сохранением когерентной связи зародышей с матрицей. Вследствие этого решетка в местах сопряжений выделяющейся фазы с матри-цей находится в упр угод сформированном состоянии. Вторая стадия – выде-ление и рост (коагуляция) новой фазы (карбидов, карбонитридов) с разрывом когерентности с матрицей и снятием упругих напряжений решетки.
Снятием напряжения в связи с полным выделением карбидов, по-види-мому, и обусловлено некоторое улучшение коррозионной стойкости аустенит-ной стали при увеличении продолжительности провоцирующего нагрева (рис. 29). Есть основание полагать, что карбиды выделяются не все сразу: в то время как часть карбидов выделилась, другие находятся на стадии предвыделения, а третьи - на стадии накопления элементов. Как следует из рис. 33 полное восстановление коррозионной стойкости аустенитной и ферритно-аустенитной стали, прошедшей процесс термического старения, происходит при термичес-кой обработке (нагрев выше 900 до 1100° С, быстрое охлаждение), обеспечи-вающей растворение и фиксирование углерода в твердом растворе. Ферритные стали в этих условиях, наоборот, становятся восприимчивыми к межкристал-литной коррозии.
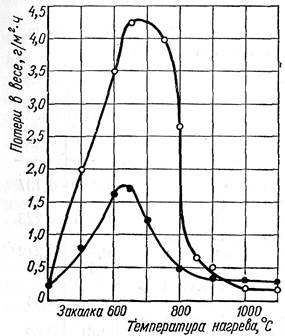
Рис. 33. Влияние температуры одночасового нагрева стали 12Х18Н10Т (верхняя кривая) и 08X21Н5Т (нижняя кривая) на скорость коррозии в
56% -ной кипящей азотной кислоте.

